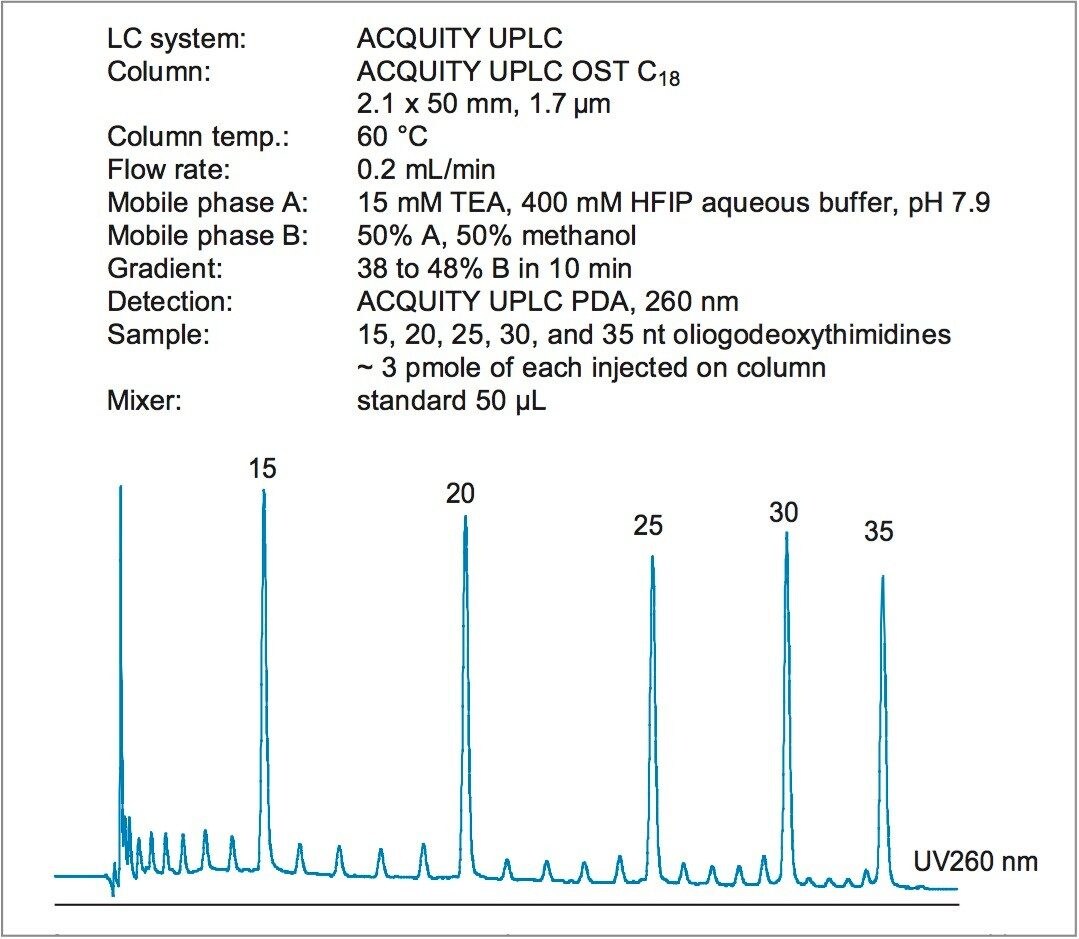
Phosphorothioate oligonucleotides are more difficult to analyze than phosphorodiester ones. When replacing an oxygen atom in the oligo backbone for sulfur, multiple diastereomers are created. Partial separation of isomers broadens the peaks in both capillary electrophoresis (CE) and liquid chromatography (LC), and complicates the analysis.
While the traditional triethylammonium acetate (TEAA) ion-pairing system is useful for phosphorodiester oligonucleotides, it fails when applied for separation of phosphorothioate oligonucleotides. Recently, Fountain and Gilar described a novel ion-pairing buffer suitable for efficient analysis of therapeutic phosphorothioate oligonucleotides.1,2 The buffer is comprised of triethylamine (TEA, an ion-pairing agent) and aqueous hexafluoroisopropanol (HFIP, a volatile weak acid used as buffering component to bring the pH to ~8). In addition, this ion-pairing system is compatible with both UV and electrospray MS detection.
The method development for oligonucleotide separation includes an optimization of gradient slope and initial mobile phase elution strength. The method development for analysis of modified oligonucleotides should reflect the fact that these are often more retained in IP-RP LC. An adjustment of initial mobile phase strength may be necessary, especially for 2'O-methylated oligos.
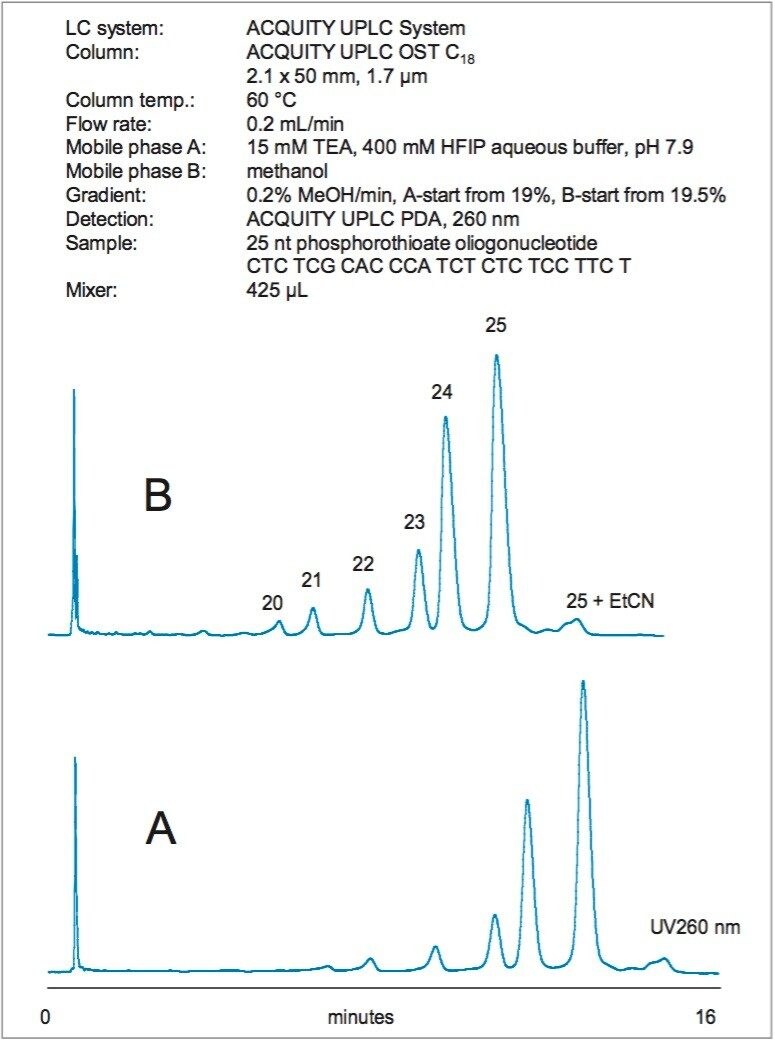
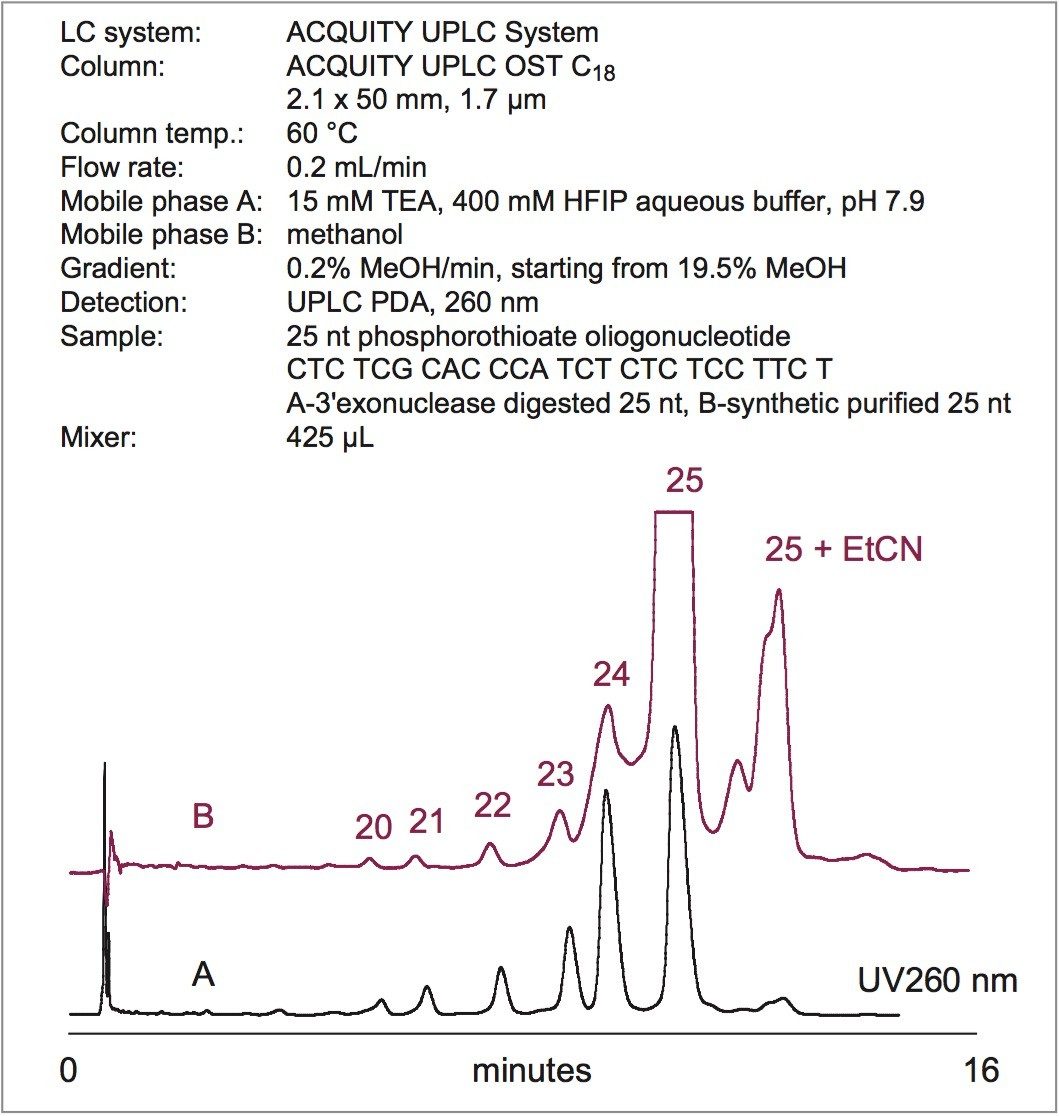
Figure 2 shows the separation of 25 nt phosphorothioate oligonucleotide that was partially hydrolyzed with snake venom phosphodiesterase (3'-exonuclease). The main 25 nt peak was clearly resolved from the N-x 3' truncated species. The identity of the peaks was confirmed by their mass (data are not shown).
The gradient slope used for phosphorothioate separation was 0.2% MeOH per minute. In order to maintain a smooth gradient profile when generating the gradient from 100% aqueous and 100% organic mobile phases, the larger mixer (425 µL) is recom-mended. Figure 2 illustrates that the analysis time can be reduced without sacrificing a resolution. This is achieved by appropriately adjusting the initial gradient strength while keeping the gradient slope constant.