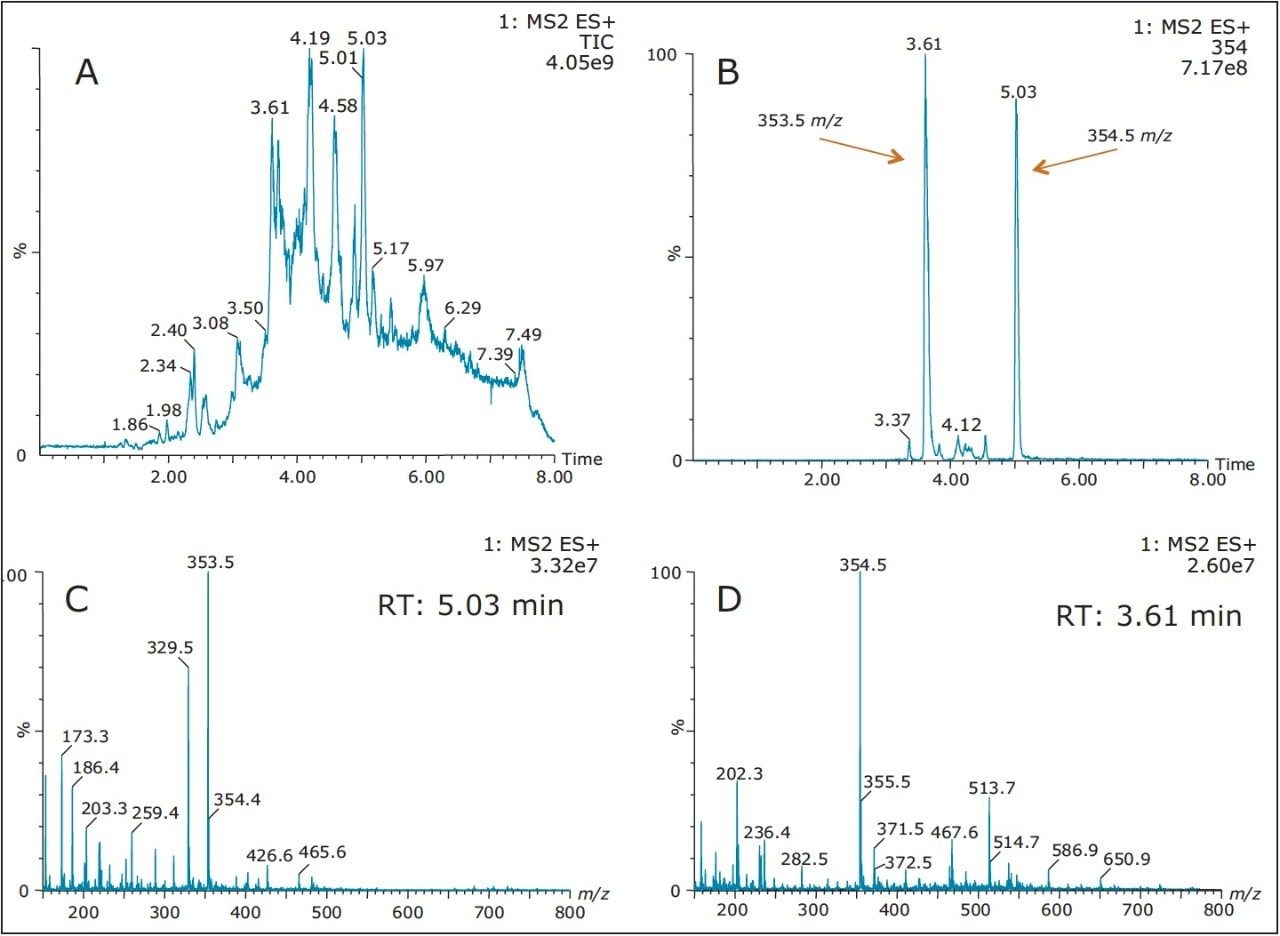
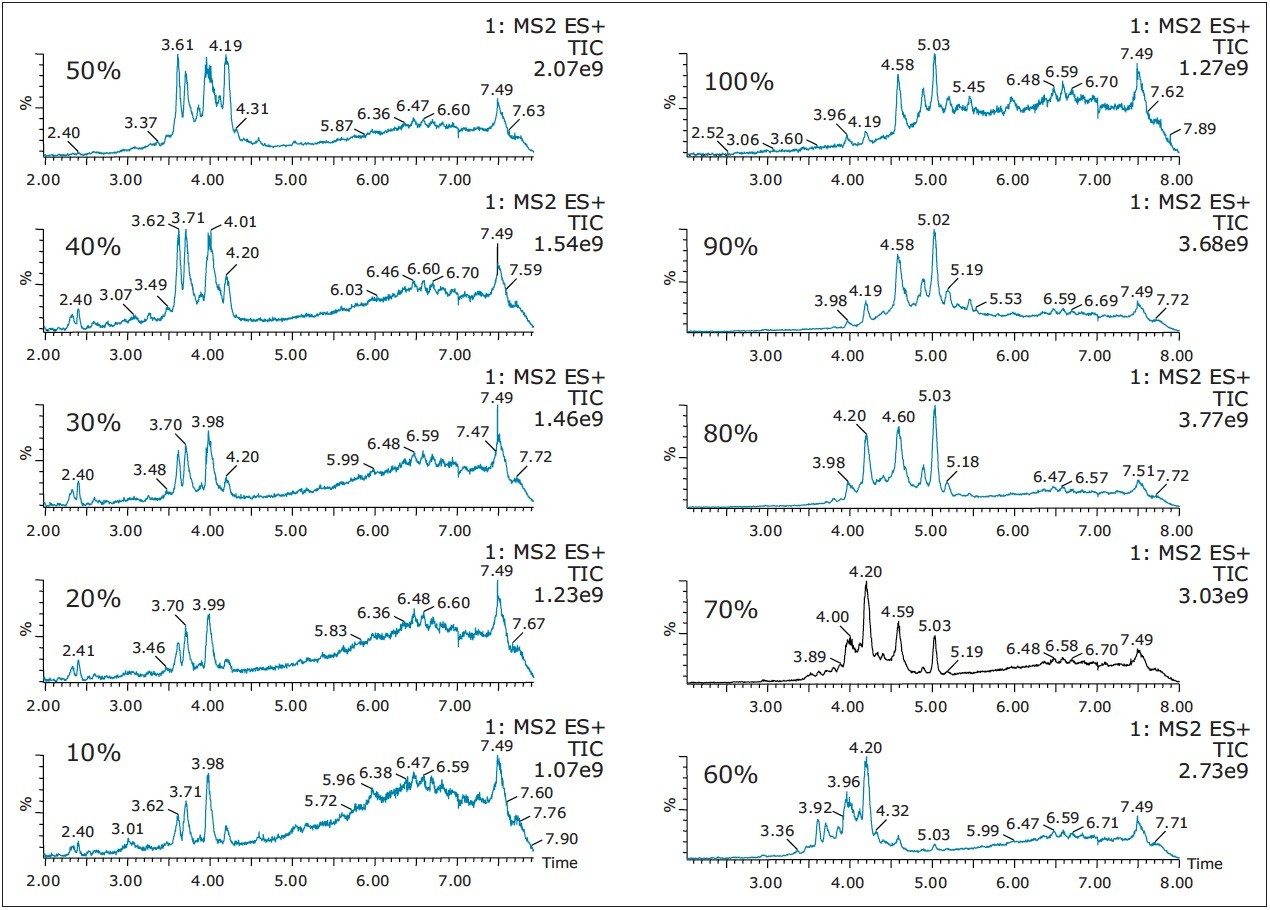
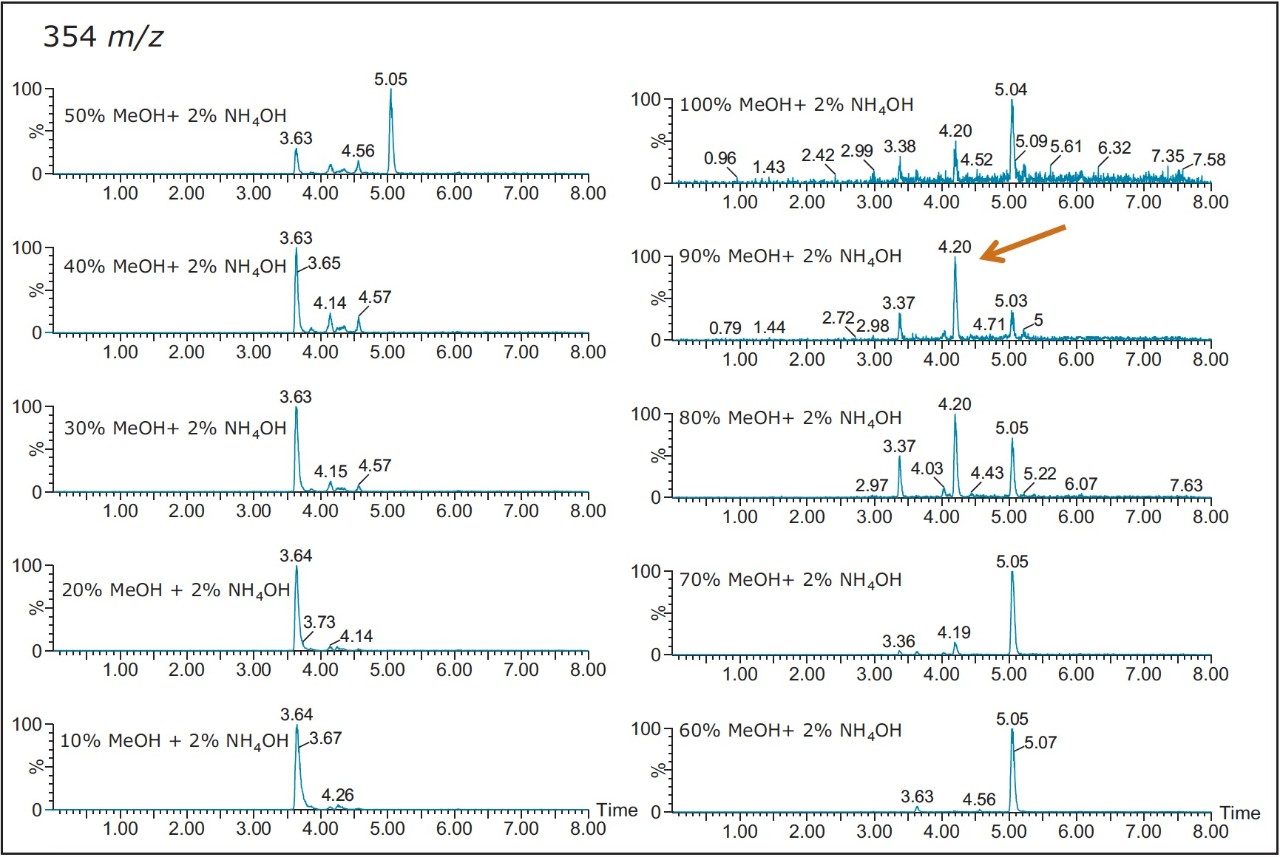
Also, at high pH elution, the 5.05 min entity shows weak solubility in acetonitrile and acetone. However, the 3.64 min entity shows a weak response with methanol. This result shows a viable option to retain and screen unknown entities based on their chemical and physical properties. Currently, most unknown impurities or metabolite identification relies on a simple solubilization step, followed by a direct injection onto an LC/MS system. The rational with this approach is related to the fear of potential breakthrough with current extraction techniques (LLE, SPE, SPME, etc.). The micro extraction process relies on a high retentive value for both hydrophilic and hydrophobic entities using a novel polymer blend (Oasis HLB) as a stationary phase. The wetting property of the co-polymer also offers high retention capabilities and is not prone to breakthrough effect by hydrophobic collapse if the sorbent is run dry. The wide pH stability (1–14) and large organic solvent compatibility enables the option of crafting various elution strategies for effective screening applications.
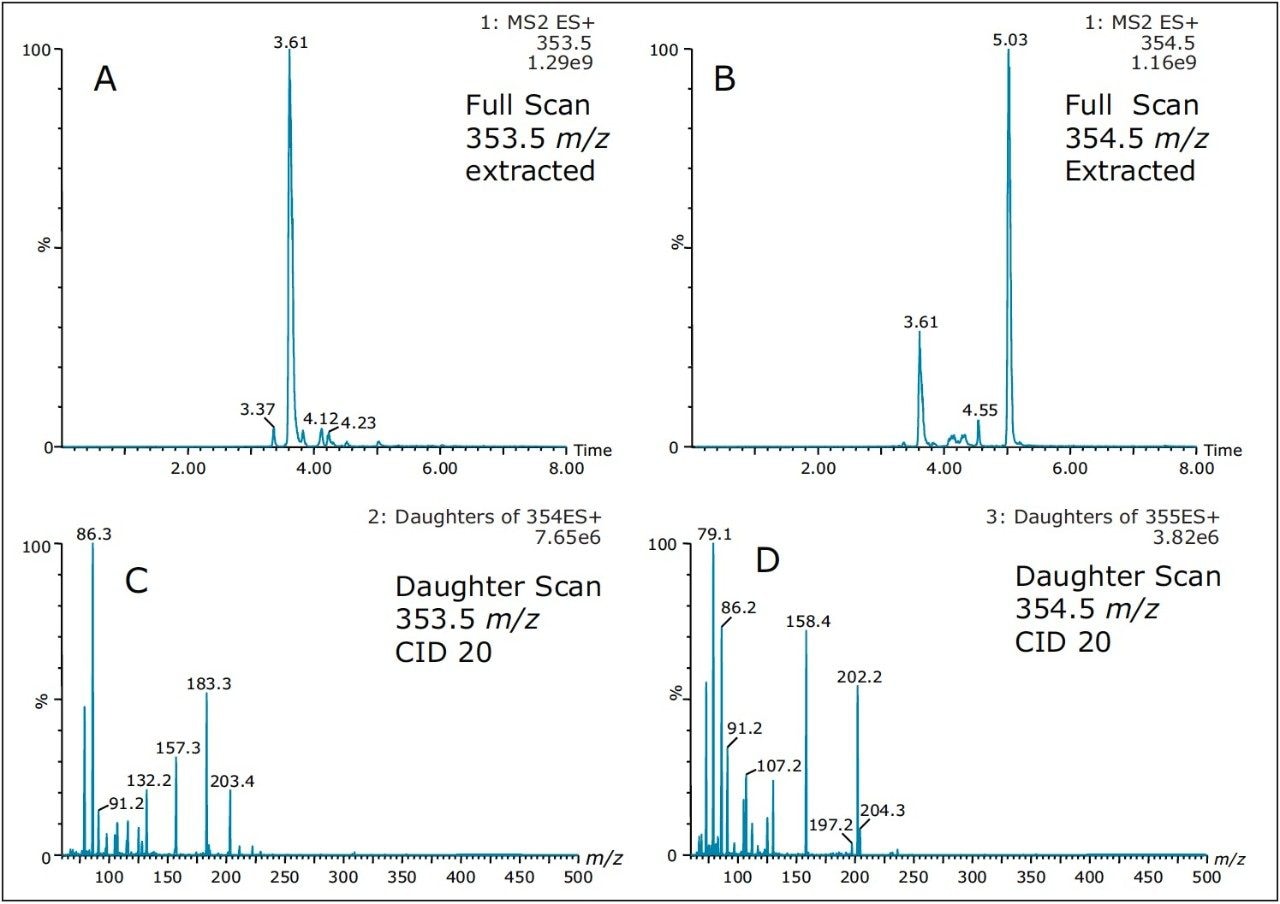
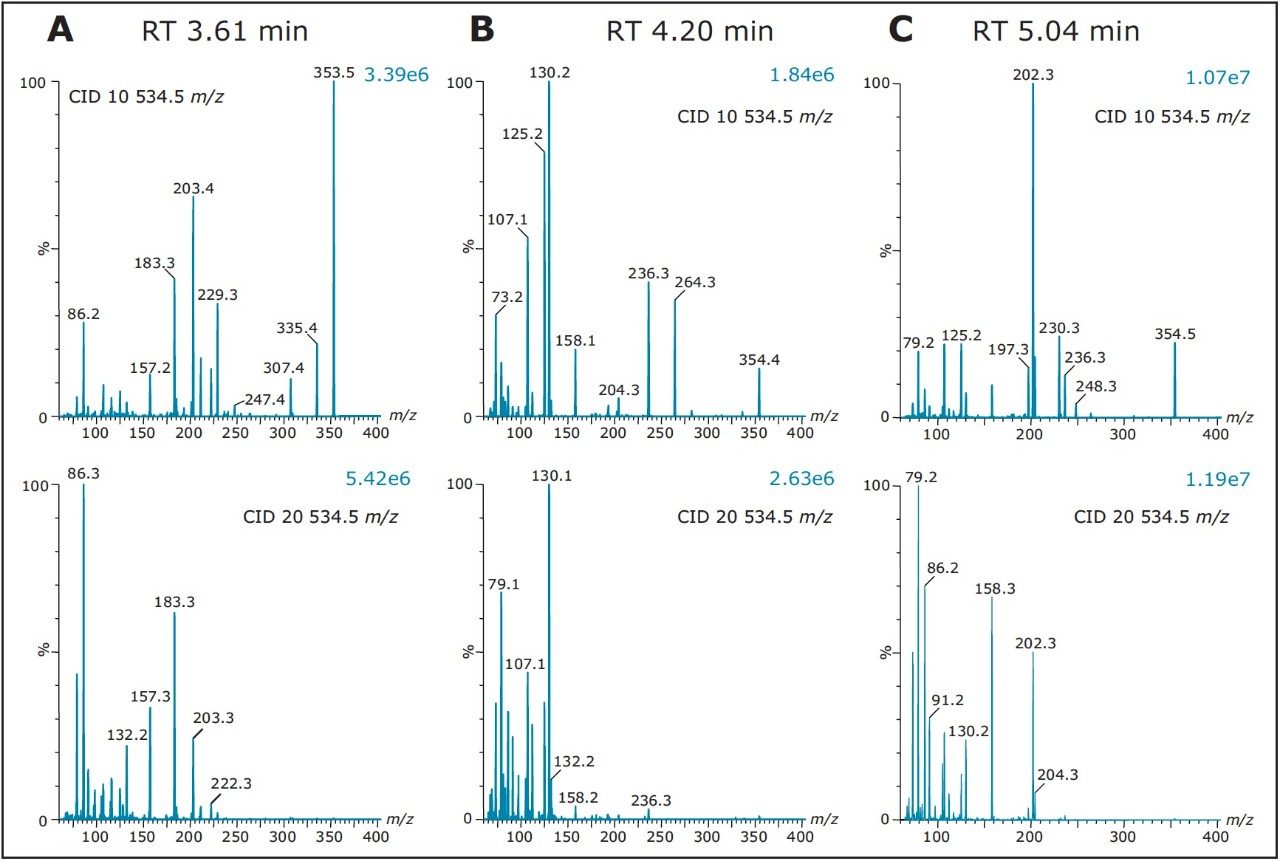
As such, Figure 8 shows product ion spectrum at 10V and 20V CID (collision induced dissociation) fragmentation values for the 3.61 min at 353.3 m/z using the maximum signal at 20% MeOH + 2% NH4OH sequential elution. The low collision value shows the parent mass at 353.3 and major fragment ions at 335.4, 229.3, 203.4, and 183.3 m/z. The product ion spectrum with the CID set at 20V shows the same common fragment ions. The strength of the micro sequential extraction is the discovery of two entities sharing the same m/z with different retention times. The entity at 4.20 min at 354.4 m/z is only detected with the high pH elution (maximum signal at 90% MeOH + 2% NH4OH) and shows major fragment ions at 264.3, 236.3, 130.2, 125.2, and 107.1 m/z. However, the entity at 5.04 min (same m/z value), show a completely different fragmentation pattern while showing a few common m/z fragmentation ions at low mass. This observation suggests the two entities share a similar backbone structure.