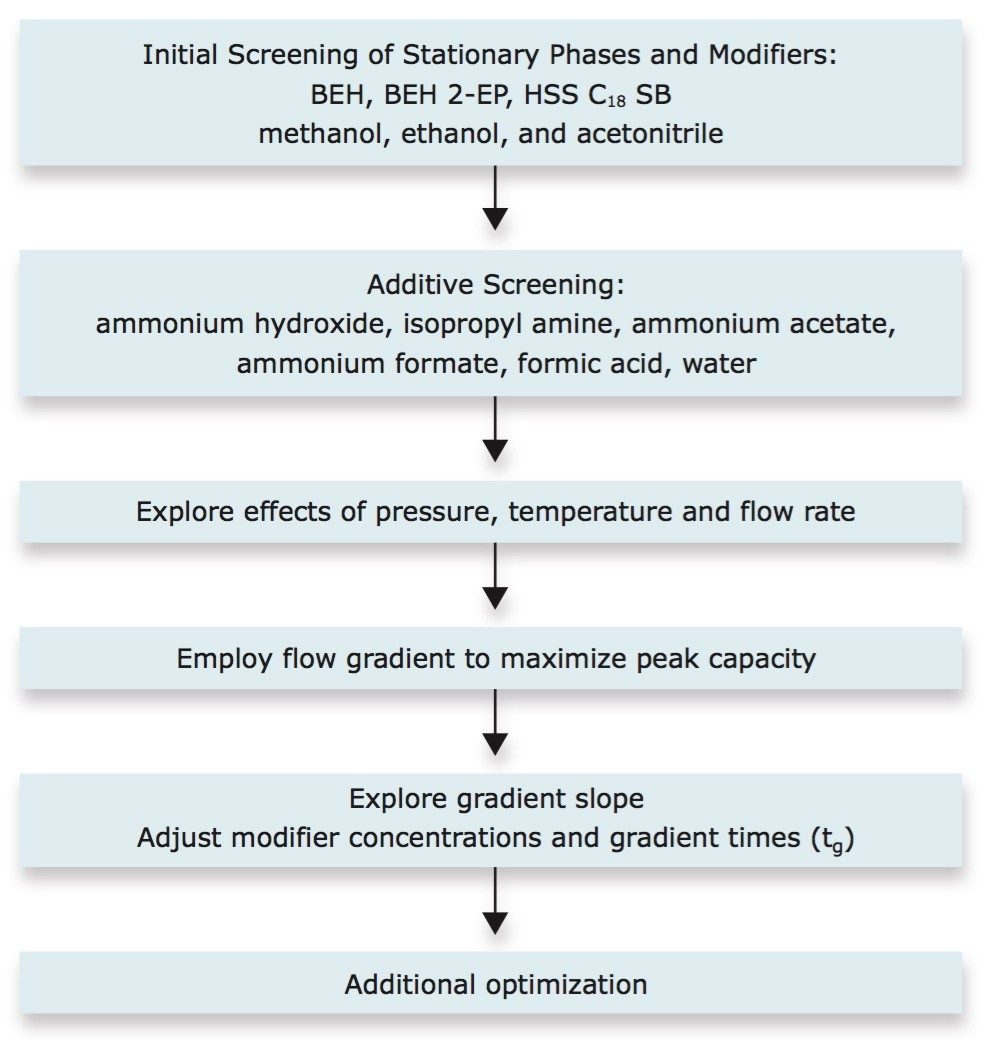
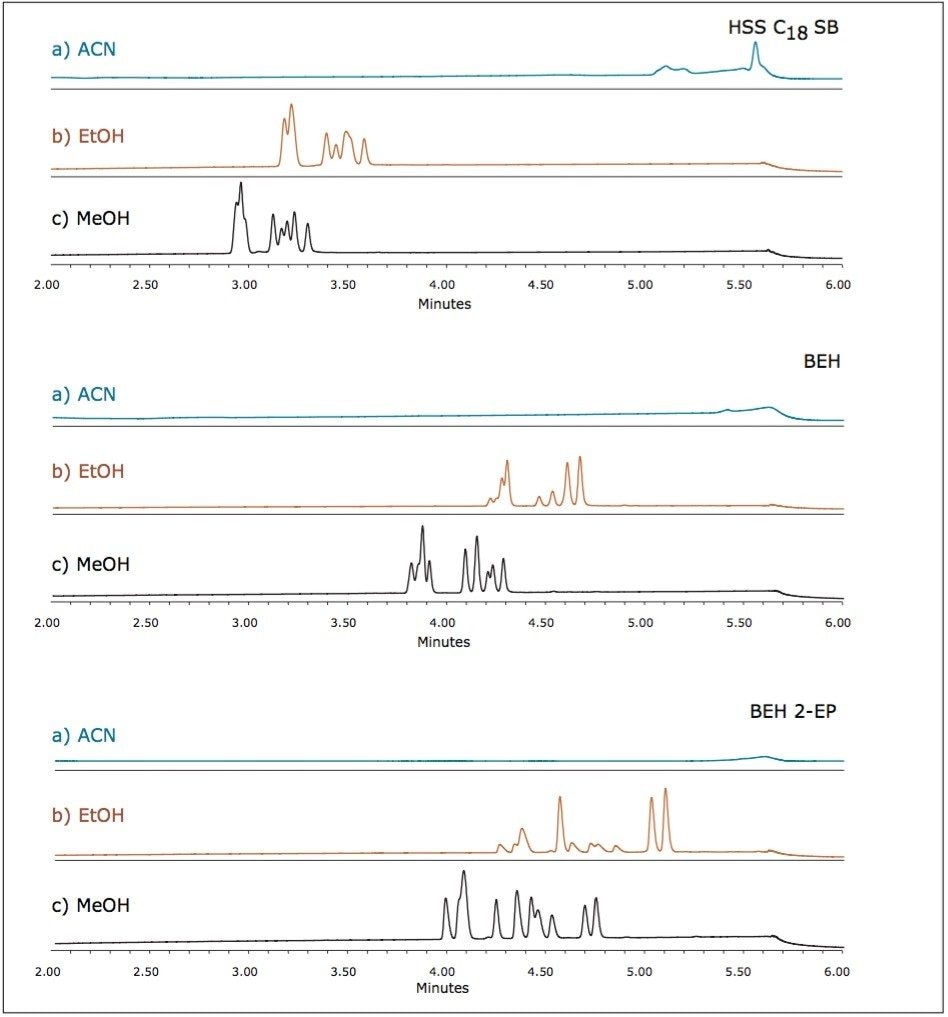
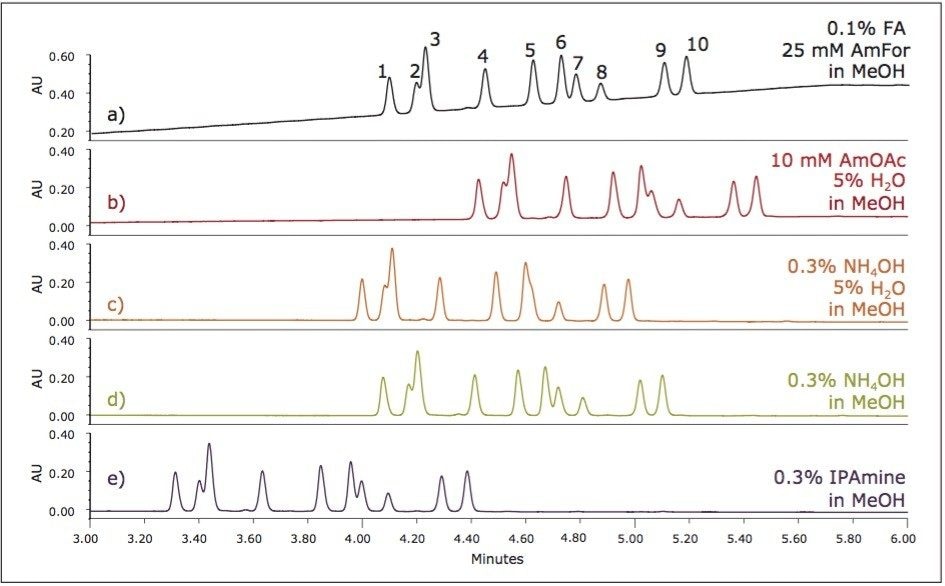
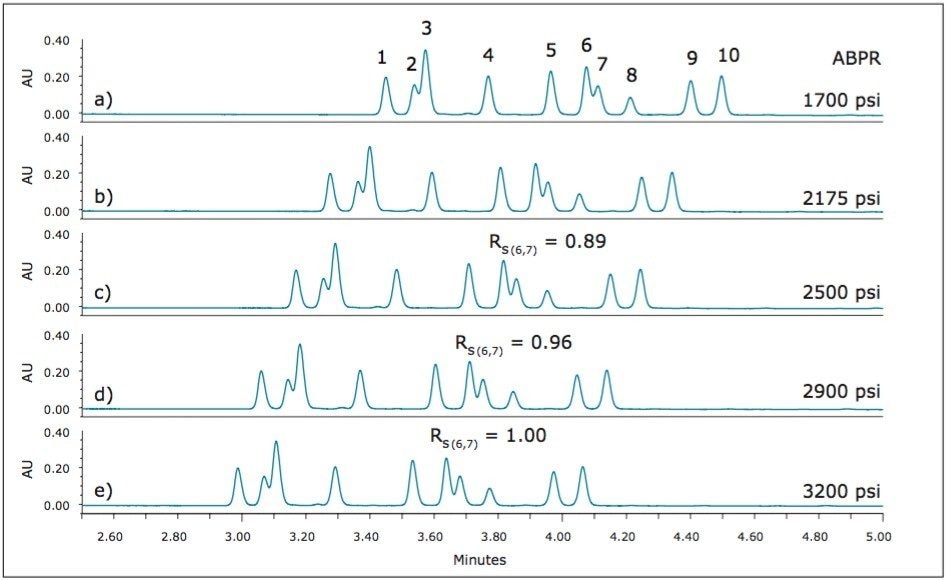
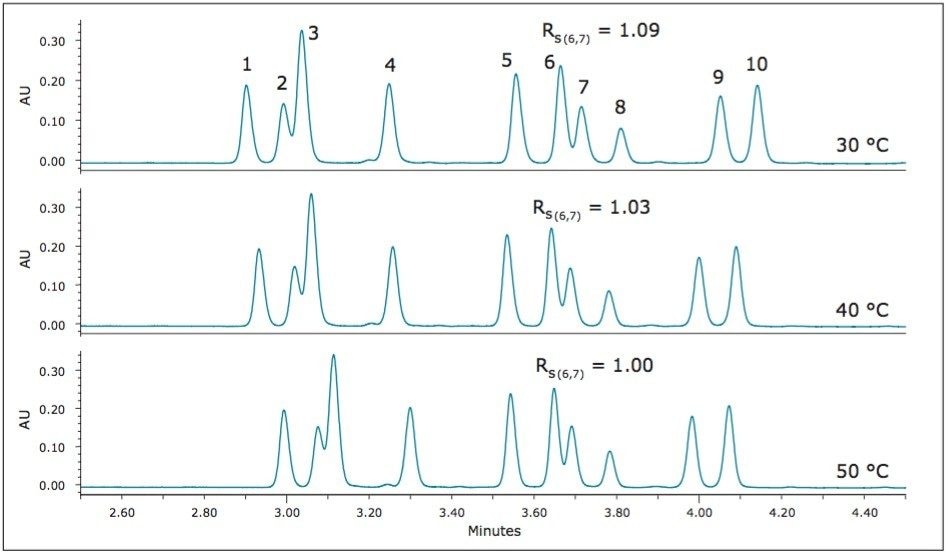
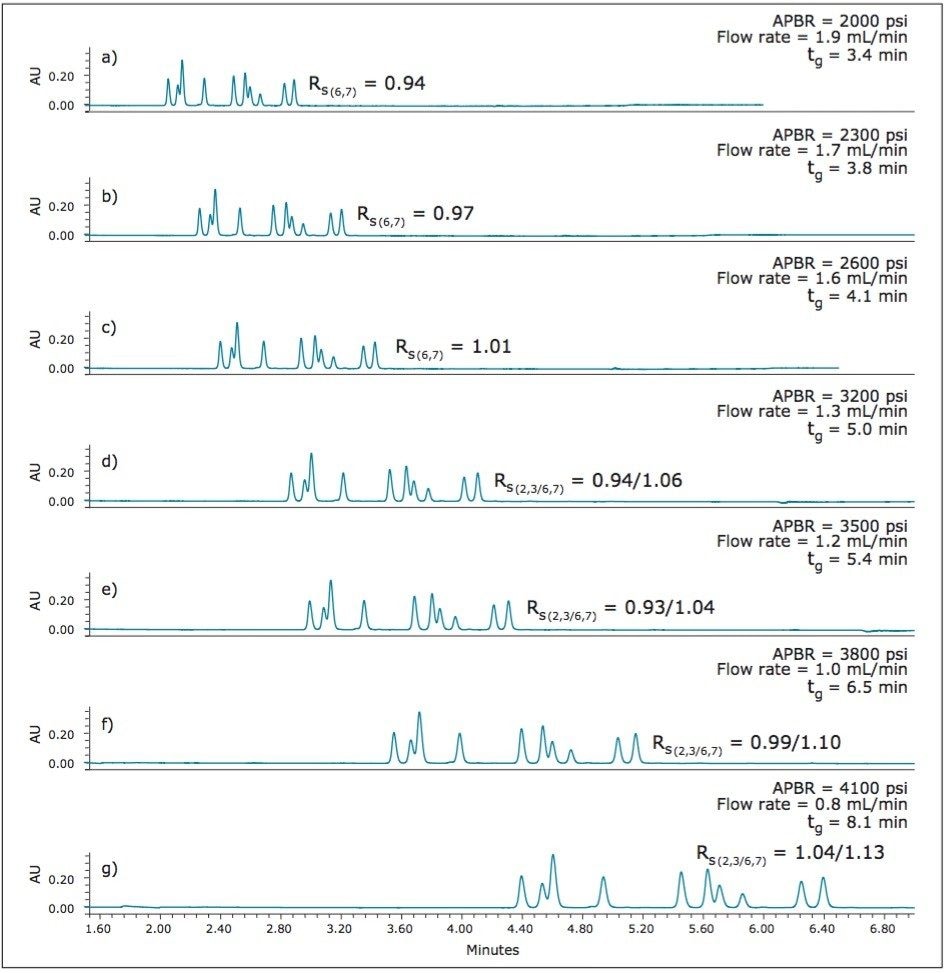
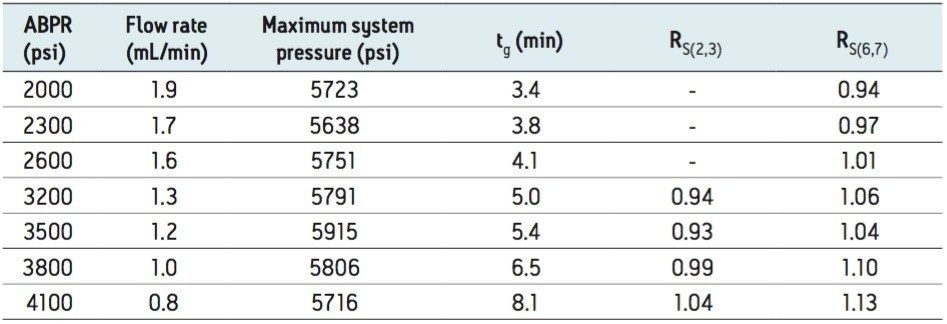
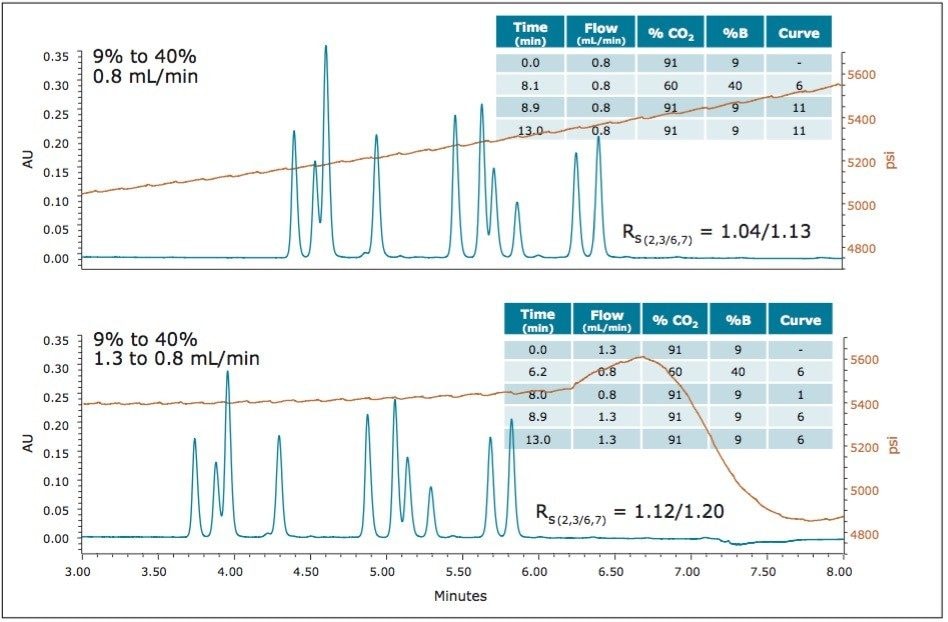
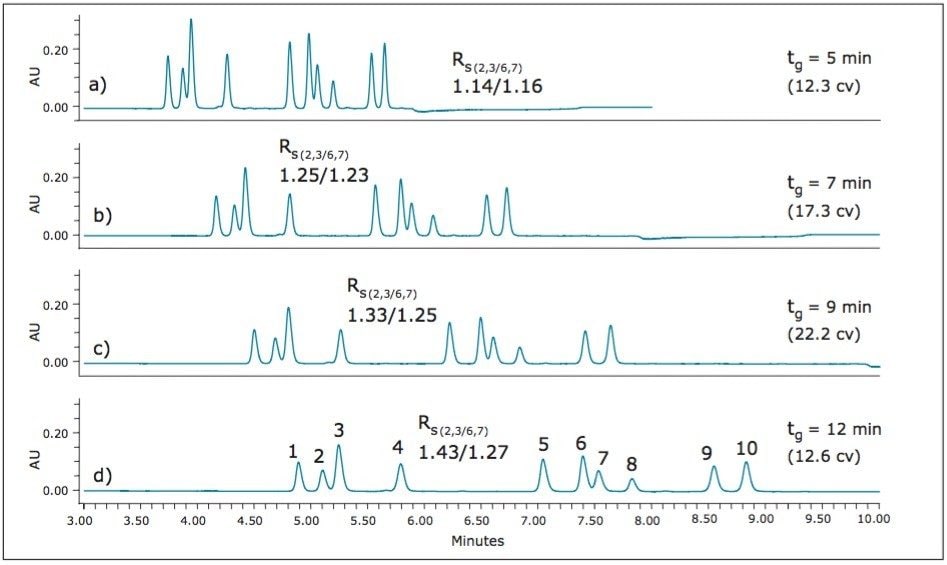
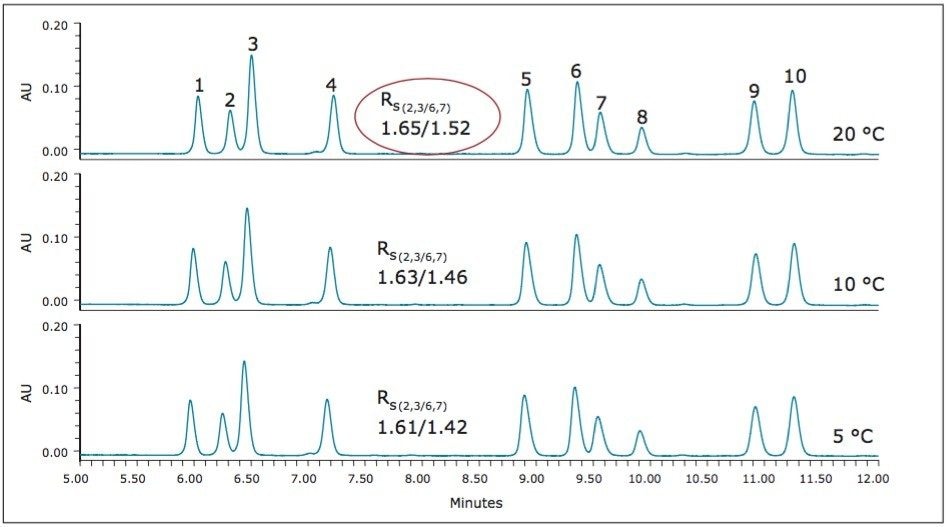
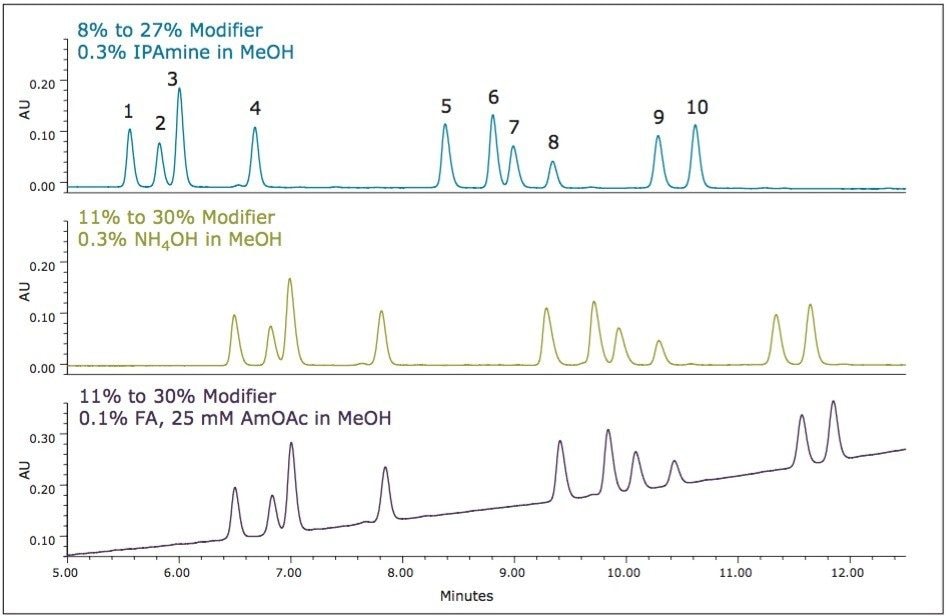
UPC2 method development for sulfated estrogens involved several critical parameters for an optimal separation, including column chemistry, choice of modifier, additive, pressure, temperature, flow rate, and gradient slope. Although the choice of additive had minimal impact on overall selectivity, the presence of the additive is critical for elution of these charged analytes, and can impact the peak shape and resolution for the analytes. The importance of pressure cannot be understated, and the application of reverse flow-gradients to maintain the highest possible flow rates and greatest efficiencies can be extremely beneficial, while staying within the pressure limitations of the system.
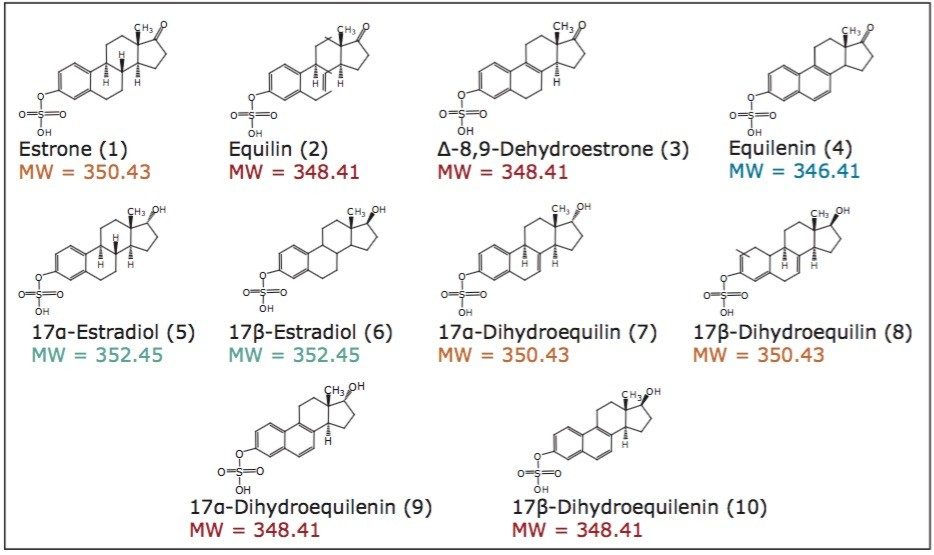
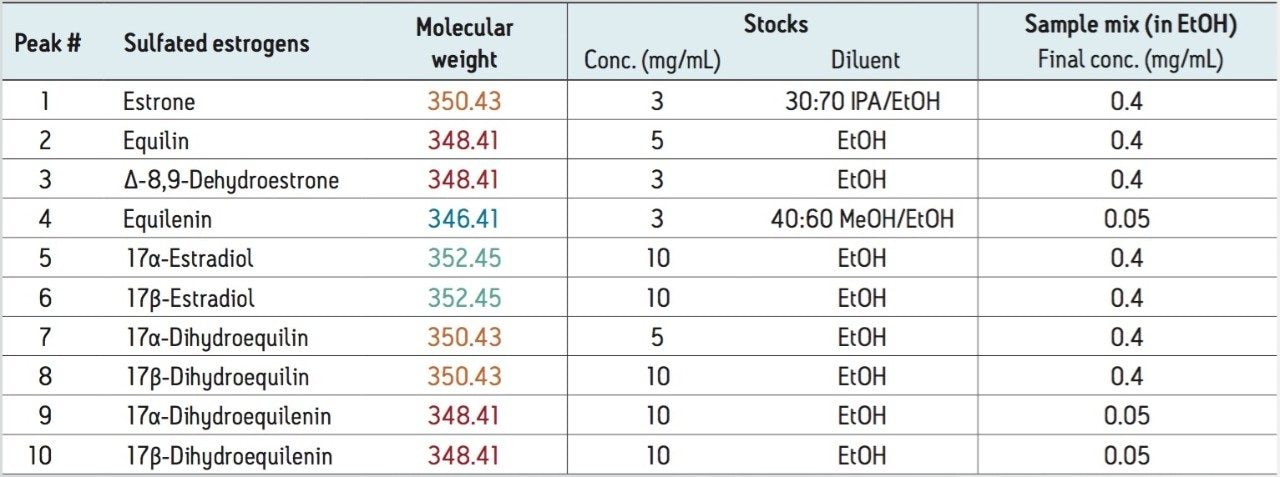
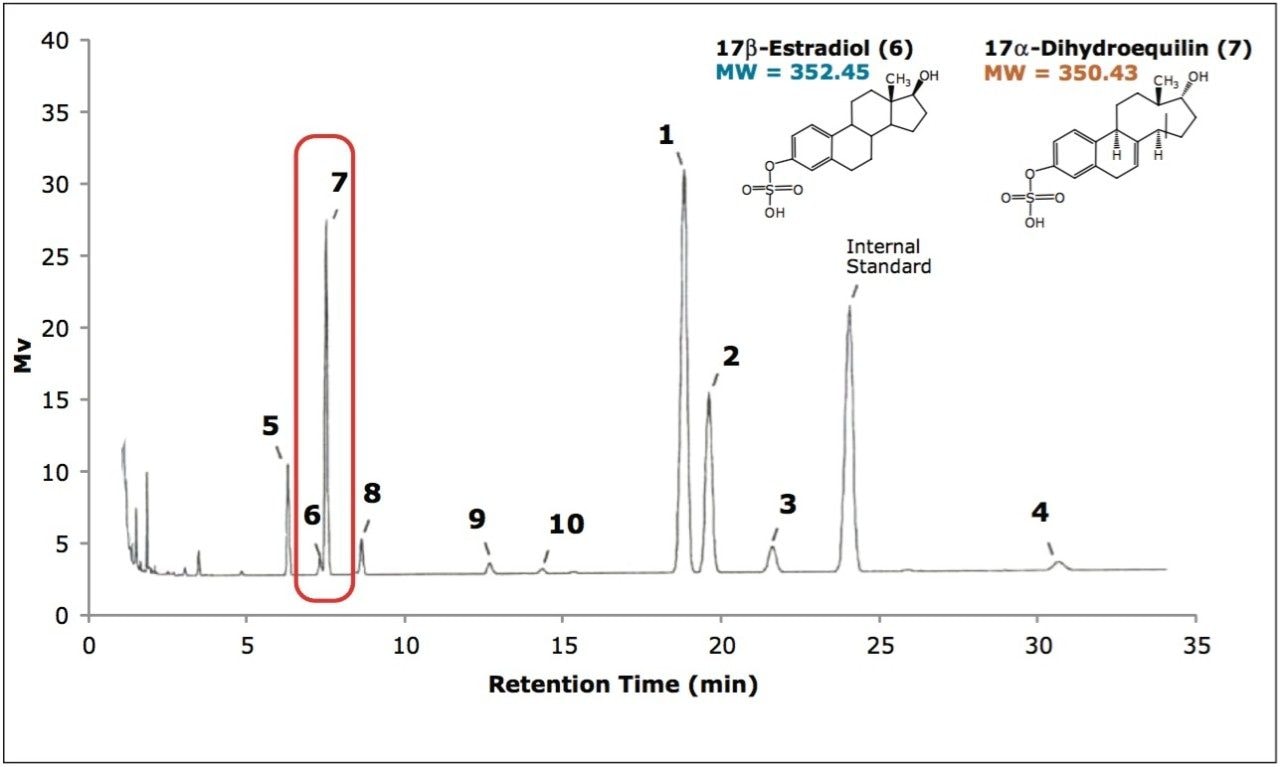
The application of this technology to the separation of sulfated estrogens represents a dramatic improvement over conventional gas chromatographic methods, with greater than 90% reduction in overall analysis time for the synthetic mixture of 10 sulfated estrogens. This is mostly due to the improvement in workflow provided by the elimination of sample desulfation and derivatization requirements prior to analysis. The reduction in the complexity and time requirements for the sample preparation yields increased laboratory efficiency with less opportunity for compounded errors. As demonstrated in this study, the ability of UPC2 technology to separate compounds with very similar structures make it well suited for the analysis of steroid and steroid-related compounds.