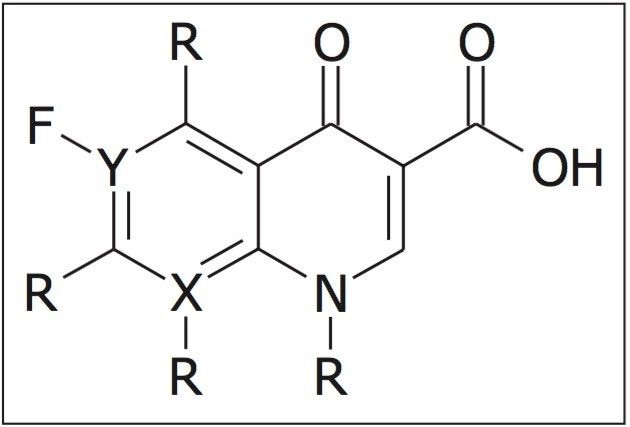
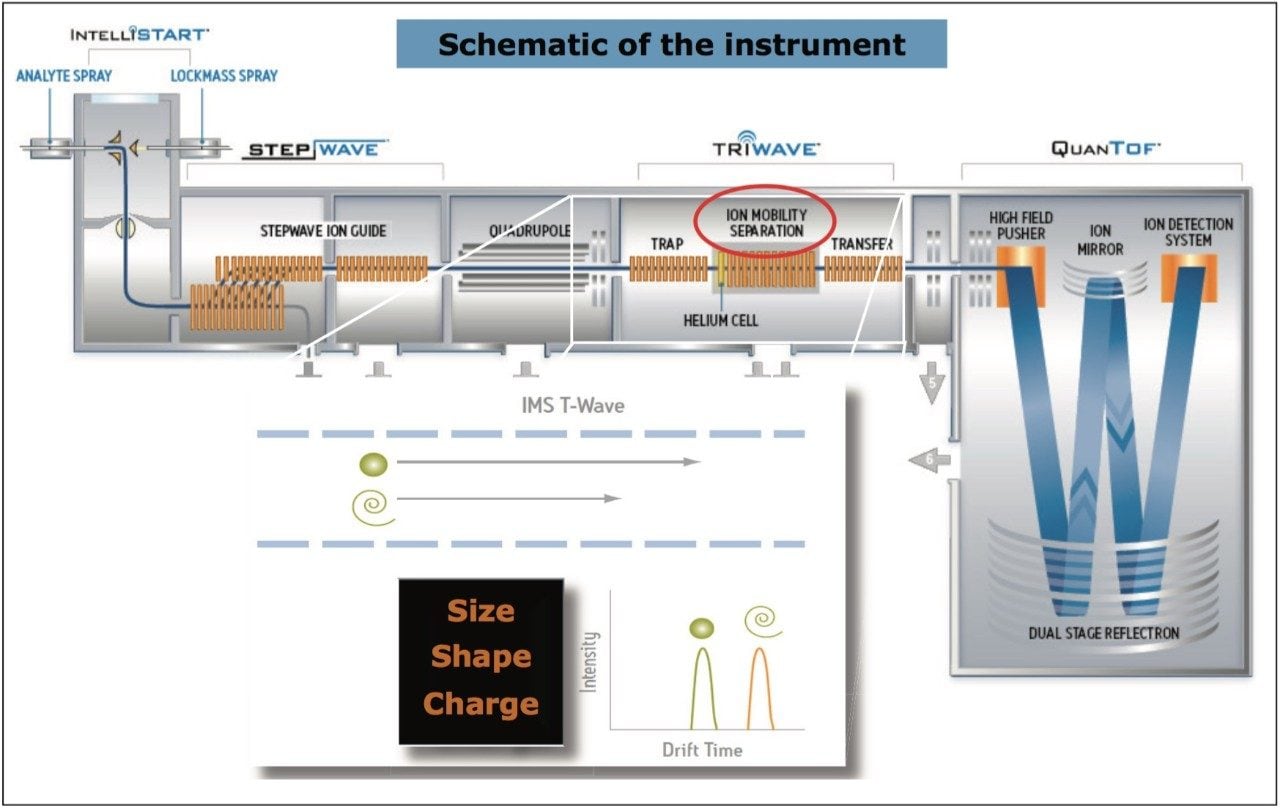
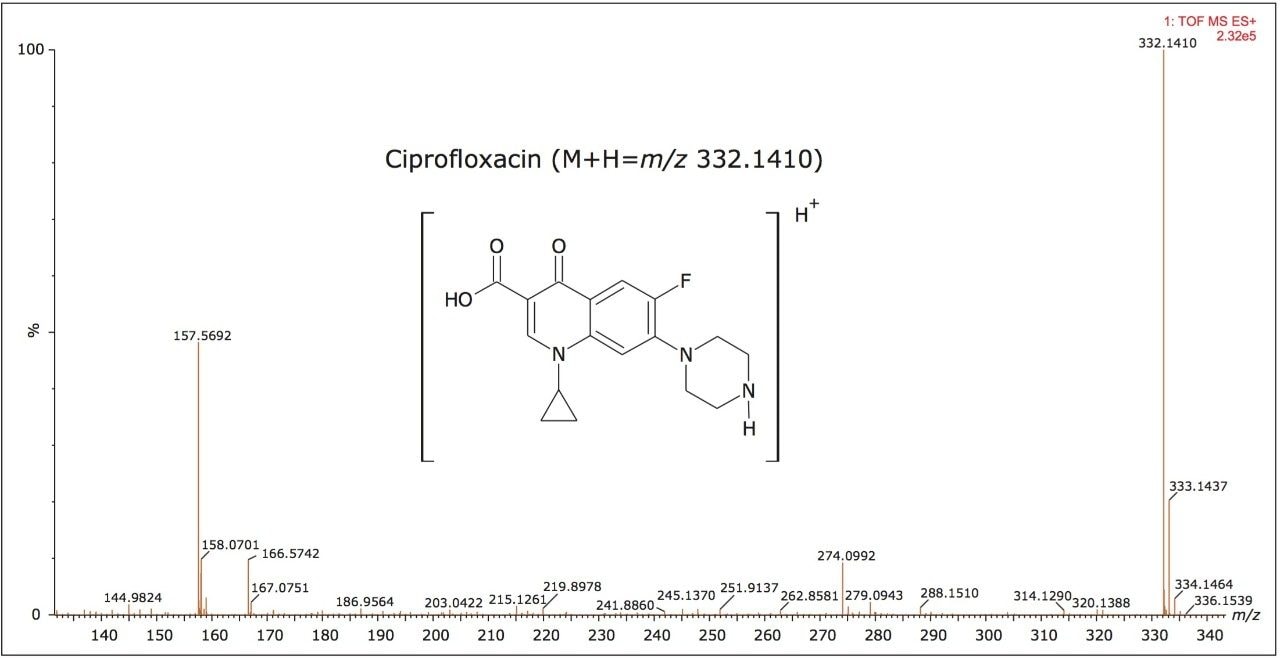
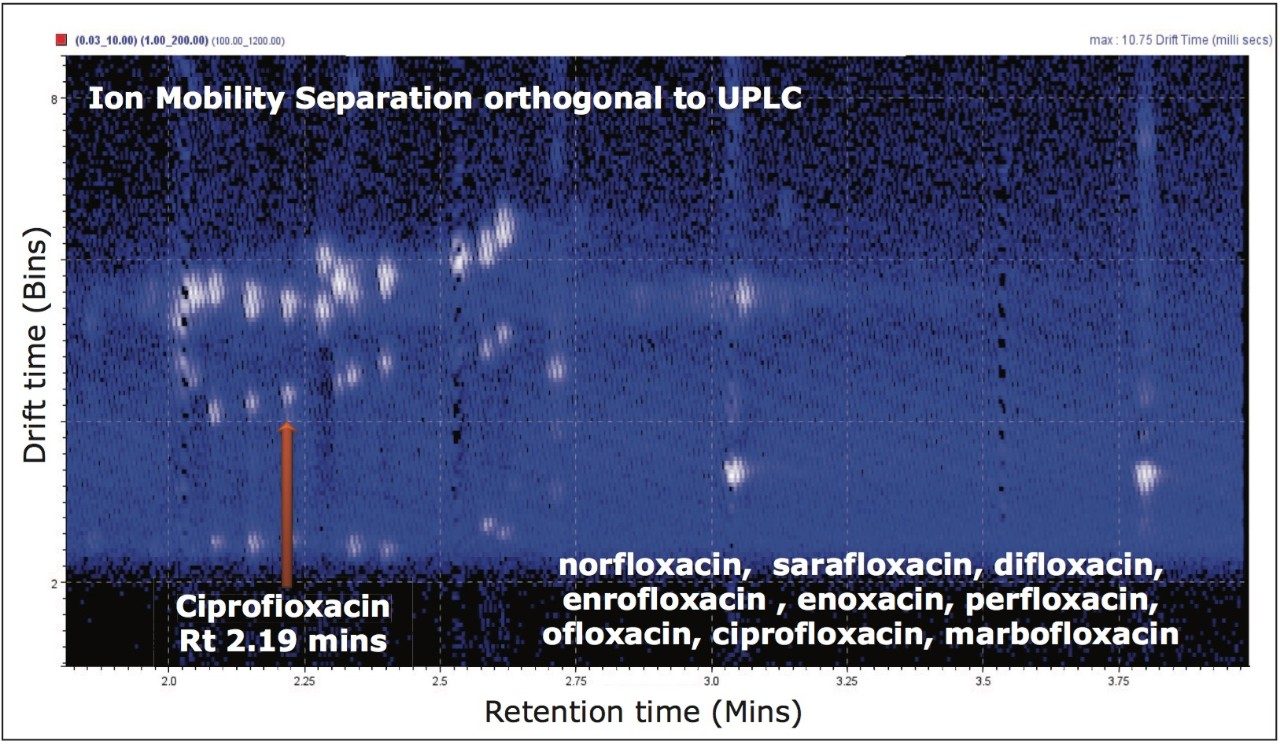
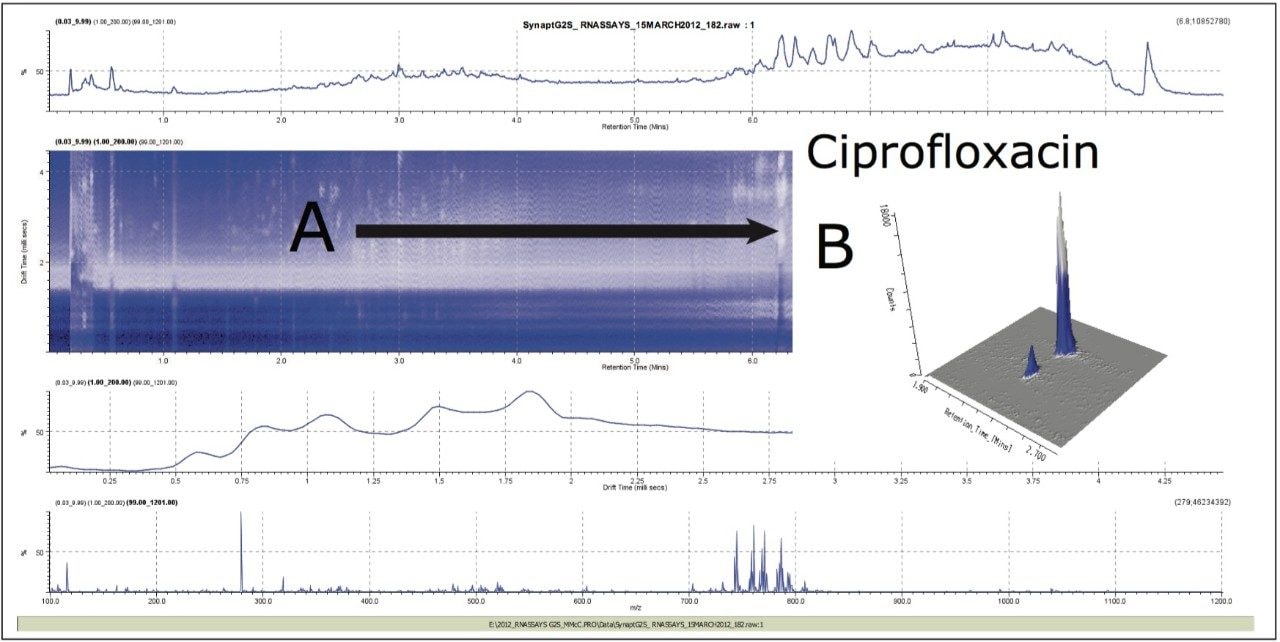
In Figure 4, the conventional MS spectrum generated in the UPLC HDMSE experiment is shown. At m/z 157 and m/z 166, two small, doubly charged ions of ciprofloxacin are apparent, indicating that ciprofloxacin did indeed also form a doubly charged species. Using ion mobility, it has been possible to separate and confirm that this small molecule does form a doubly charged species. The formation of the doubly charged species is dependent on the MS parameters used, especially the cone voltage. If the cone voltage is too high, the doubly charged species is not observed, only the [M+H]+.
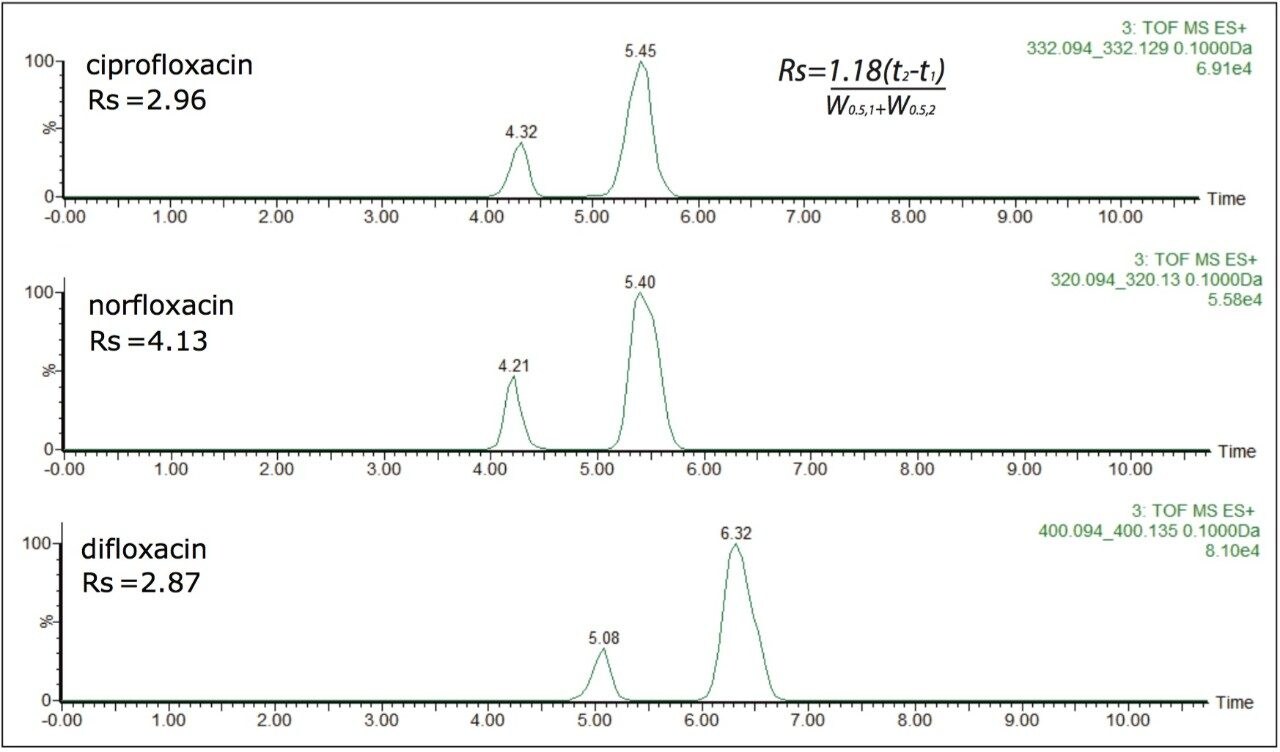
The data confirm that further consideration should be given to method development and the means of analysis chosen, since the ratio and formation of the protomers varies with eluent flow rate, capillary voltage, cone voltage, and matrix. If MRM is the method of choice, consideration of the experimental conditions used and the specific transitions selected is imperative. The data illustrate that consistency in MRM transitions in inter- and intra-laboratory studies could easily be misinterpreted within and between different laboratories, explaining the challenges of achieving reproducible results for these types of compounds. Ion mobility is a valuable tool for method development to ensure method robustness and consistent results.
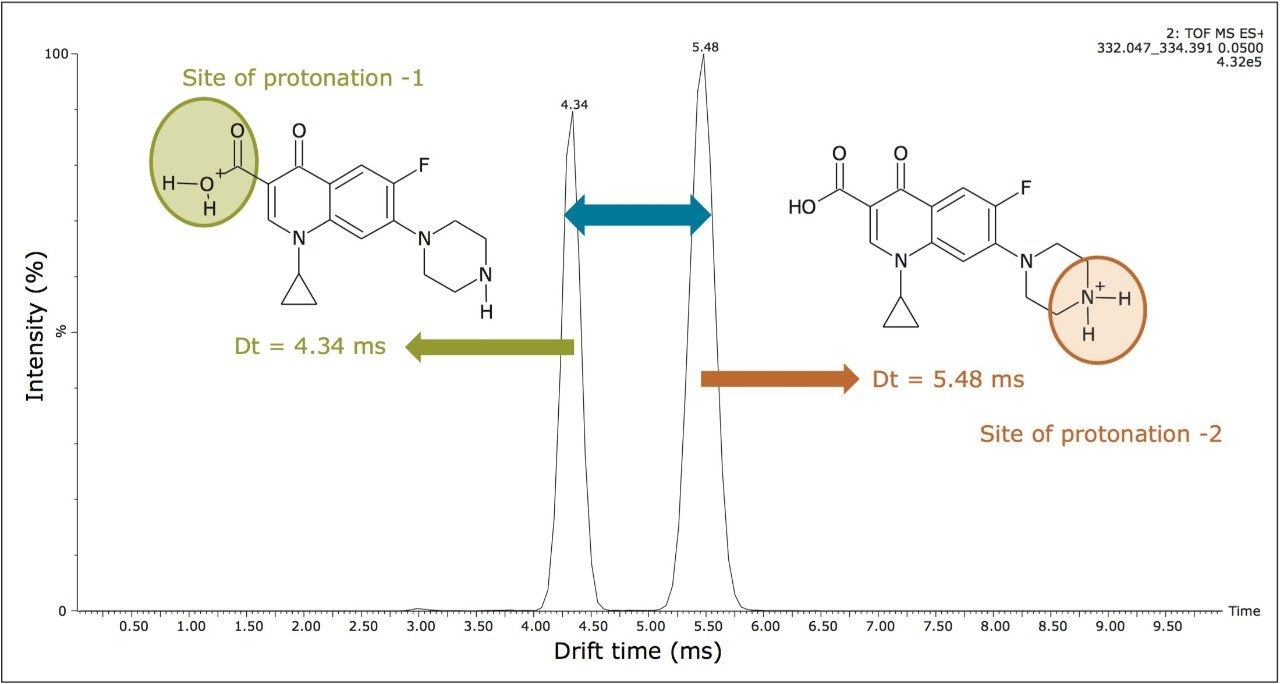
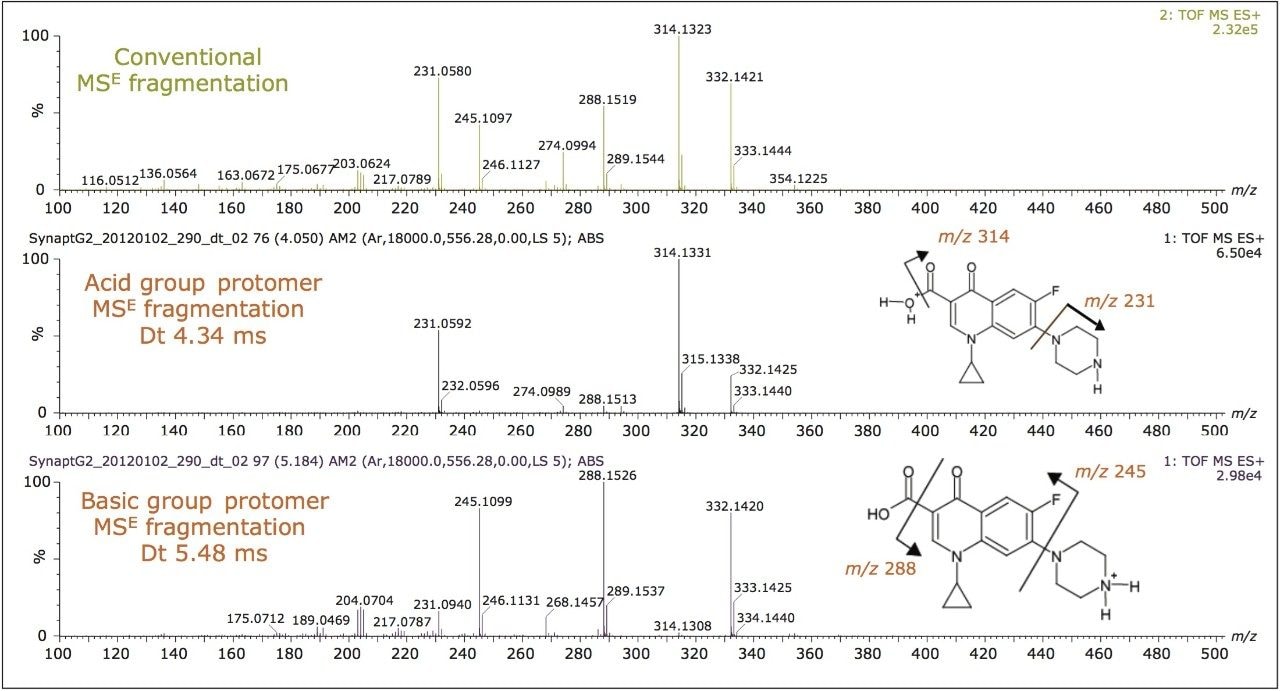
In addition to the scope of more specific and reliable method development, the drift times generated for the components analyzed can be used to produce an additional identification point. This application note described retention time, precursor ion accurate mass measurement, fragment ion accurate mass measurement, and two drift time values can be used as identification points for ciprofloxacin.
In addition to utilizing ion mobility to provide a new identification point, the orthogonal separation produced by ion mobility can be used for spectral cleanup. The plot of drift time versus retention time (Figure 8A) shows the ion intensity, represented by white pixels. The extent of the presence of matrix ions is shown by the expansive white color. The more intense analyte and matrix components are represented by the more vibrant white spots. However, it is difficult to see the low level target analytes due to the large amount of ion intensity produced by the matrix. In Figure 8B, the ciprofloxacin protomers have been extracted from the porcine matrix. Single component MS and fragmentation spectra can be produced from this clear, selective separation resulting from the use of ion mobility. The ratio of the acidic site protomer to the basic site protomer in the porcine matrix was determined to be 5:1 under these conditions. This is affected by capillary voltage, cone voltage, probe position, flow rate, and matrix. During infusion experiments, it was possible to vary the ratio of acidic/basic protomers and even change which was the most abundant protomer.