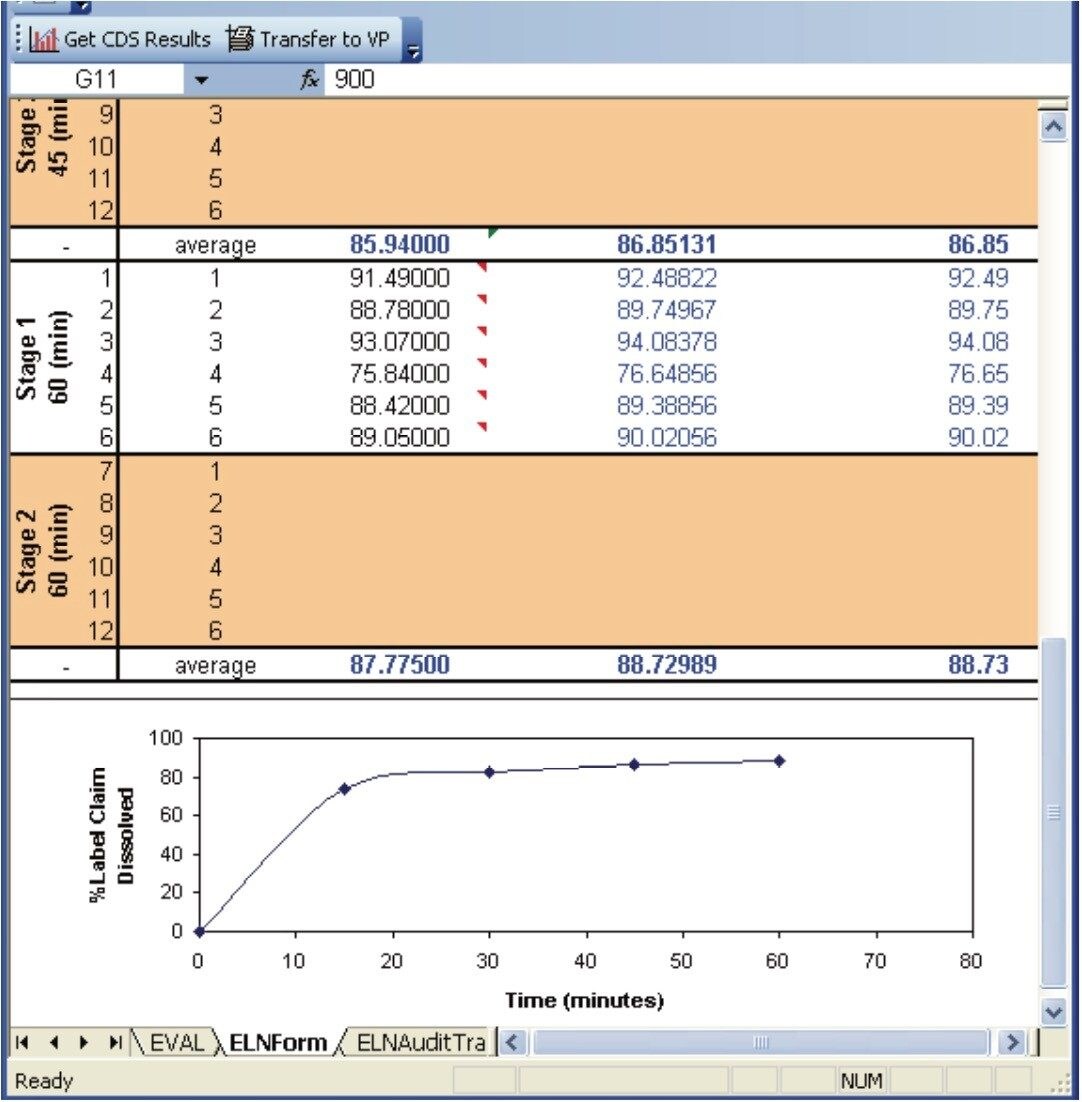
In vitro dissolution simulates in vivo drug release of solid dosage forms, transdermal patches, and suspensions.1-3 For example, in the case of tablets and capsules, the acidic environment of the stomach is emulated and the rate of drug release is measured as a function of time. Quantitative measures are made by taking small aliquots of the dissolution medium from the dissolution device at defined time points, e.g., 5, 10, 20, 30 minutes.1-3 The measurements can be performed by a number of standard analytical instruments with UV/Vis spectrometers and HPLC being some of the most common.1 In particular, HPLC methodology offers the advantages of wide dynamic range and excellent selectivity because of the chromatographic separation aspect.1
Dissolution testing may be performed during various phases of the drug development lifecycle of a dosage form, e.g., during Phases 0 and I to understand the dissolution mechanism; during Phases II and III to understand formulation; and as quality control (QC) during manufacturing. 1-2 Maintaining good documentation of the dissolution studies is a common theme no matter what the stage of the product life-cycle but is particularly important during manufacturing due to the requirement to adhere to GMP guidelines for proper procedures and documentation.
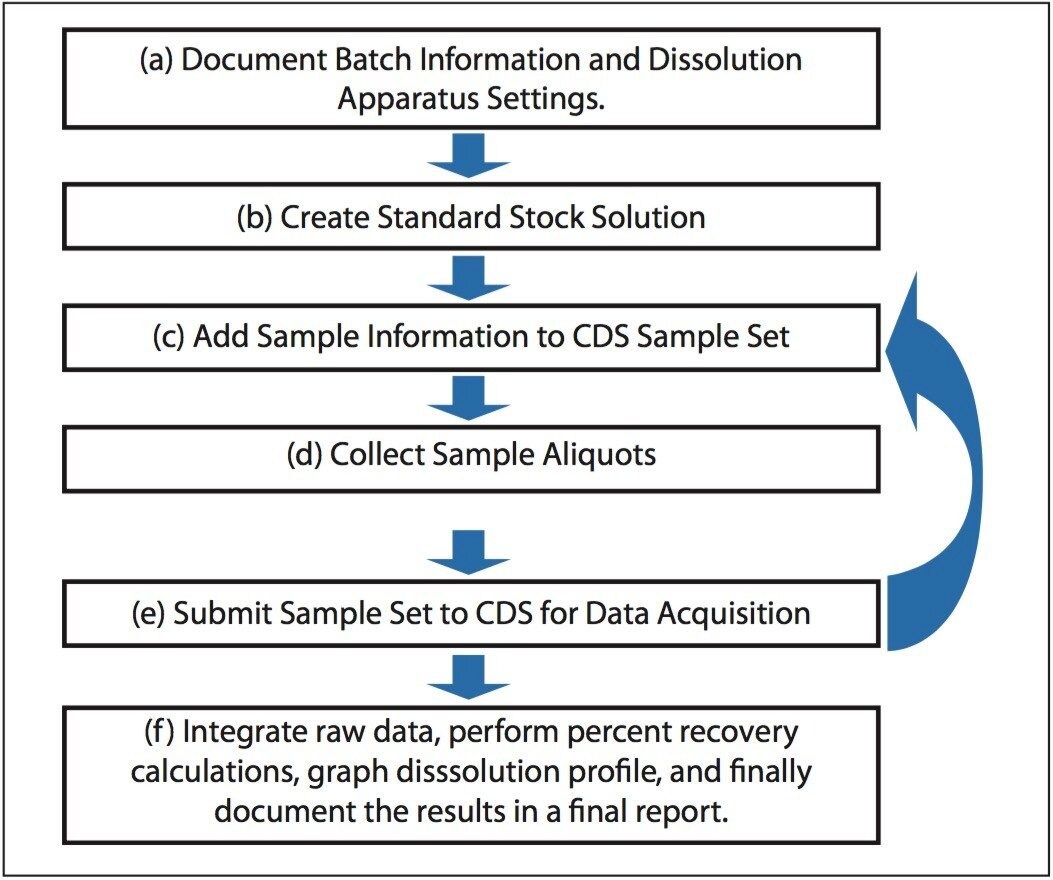
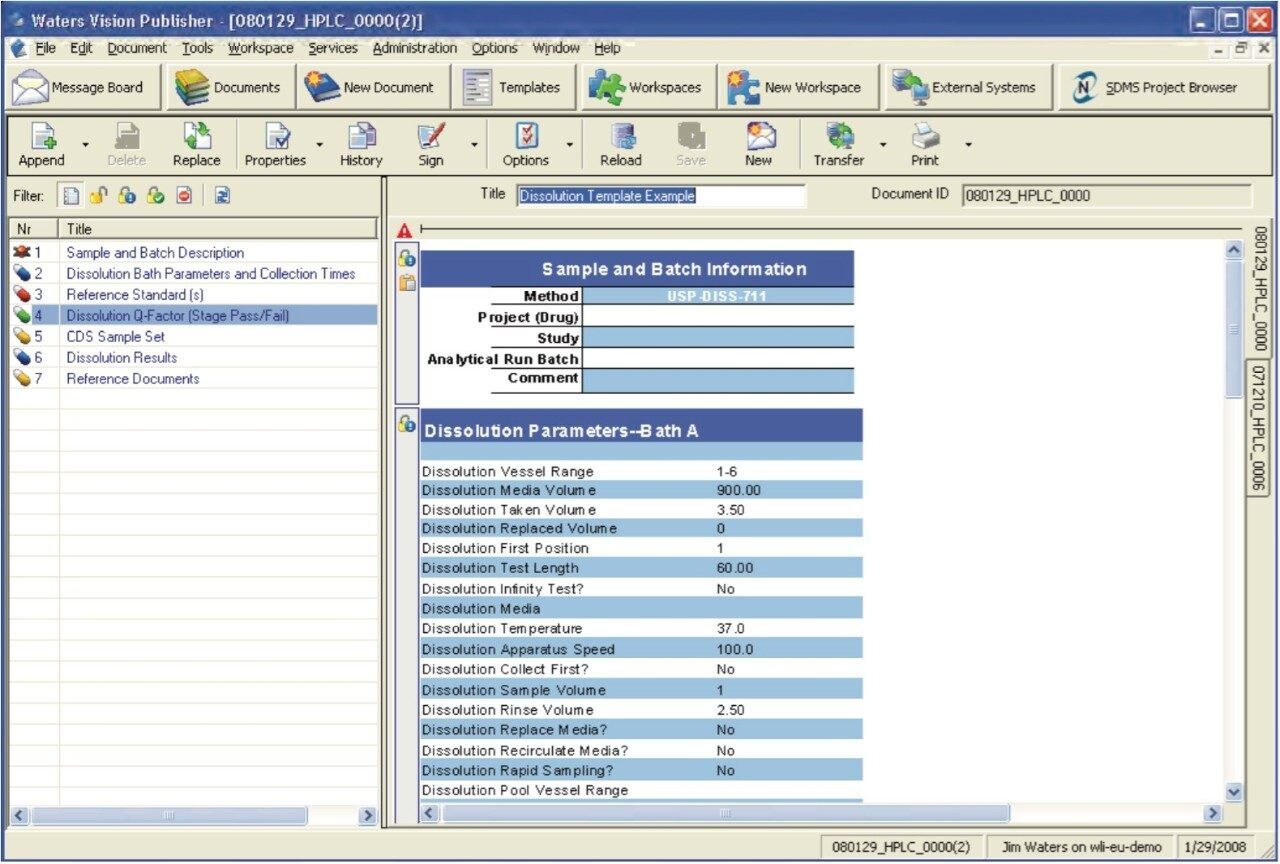
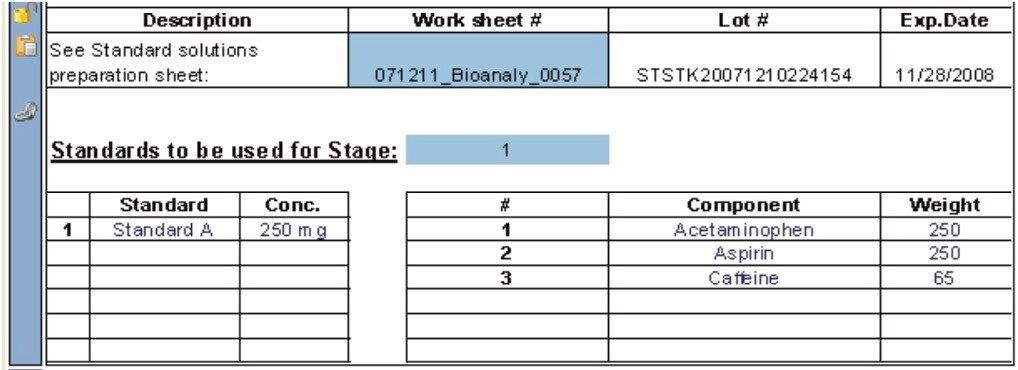
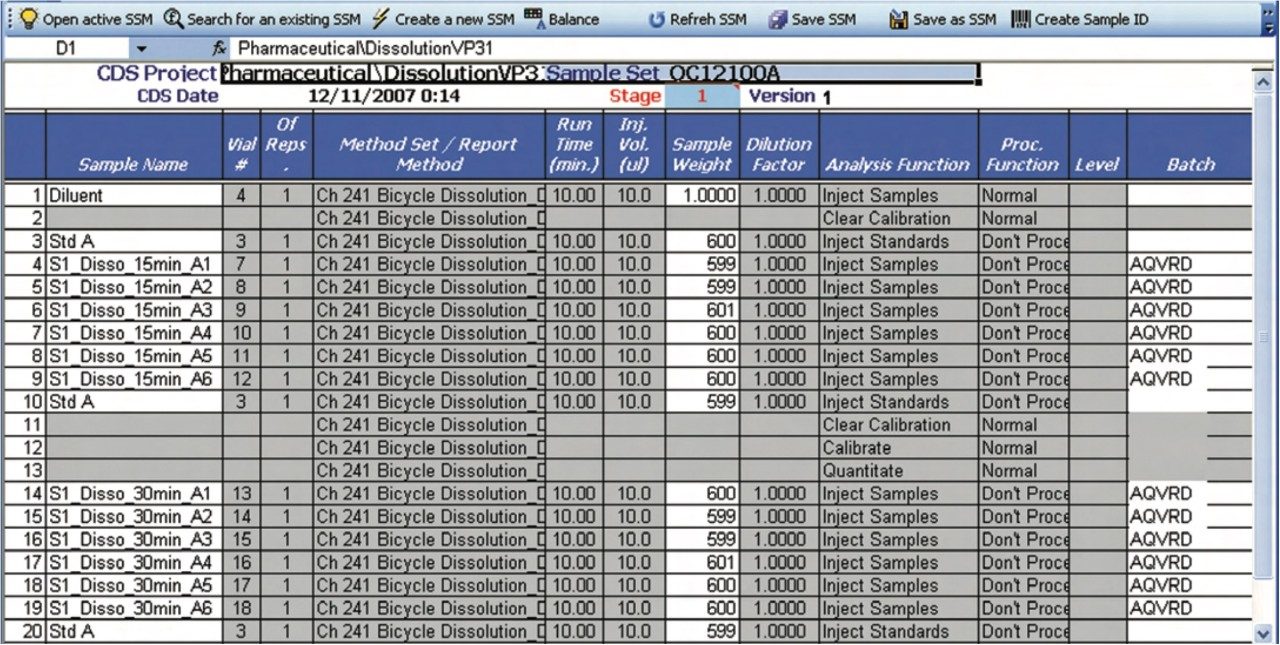
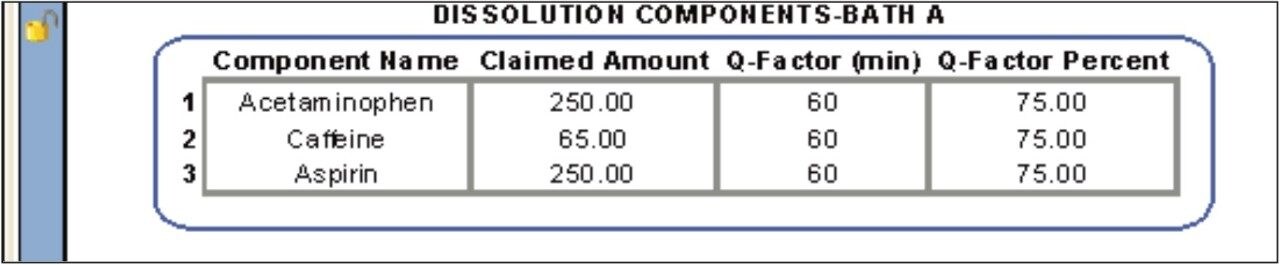
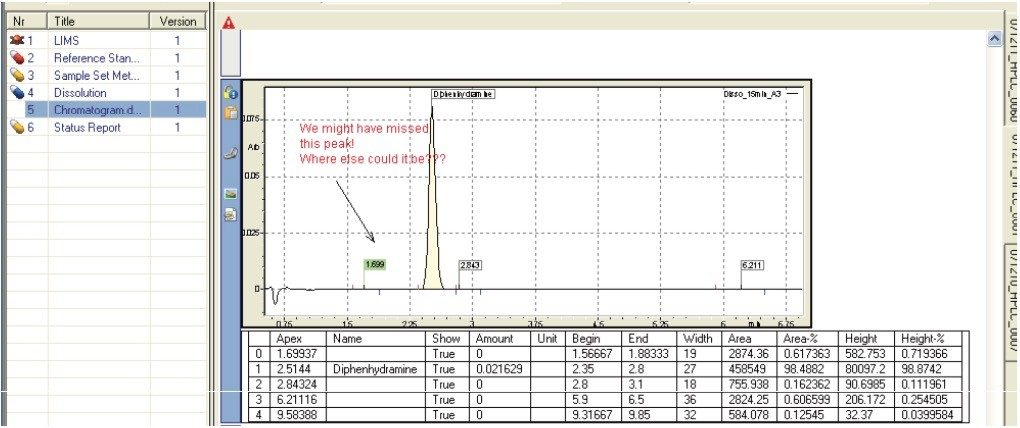
The following example, illustrates how the Dissolution Template may be used to perform a HPLC based dissolution study.