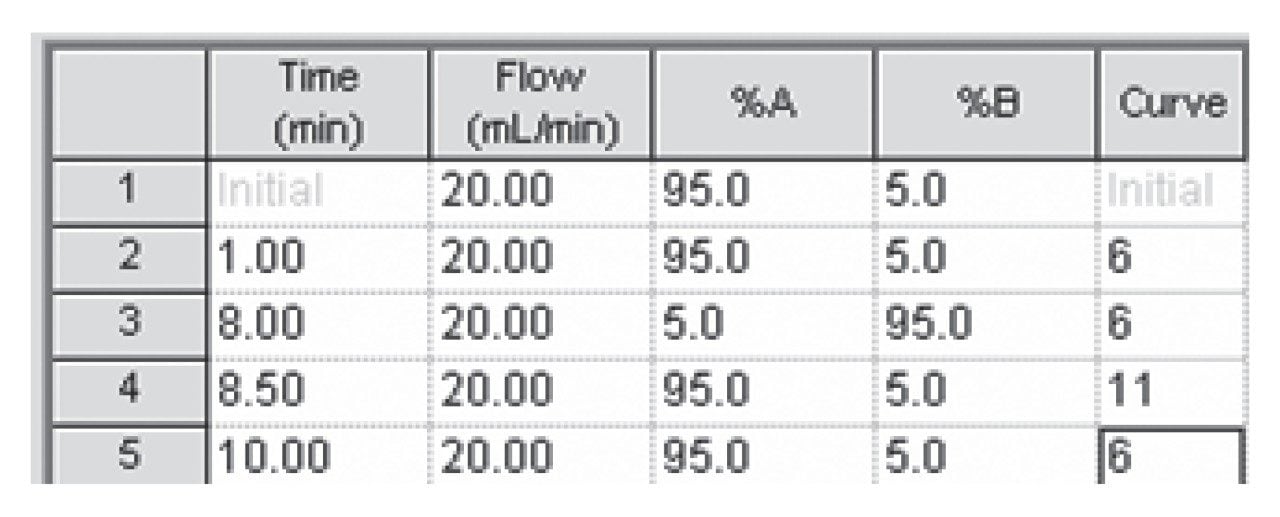
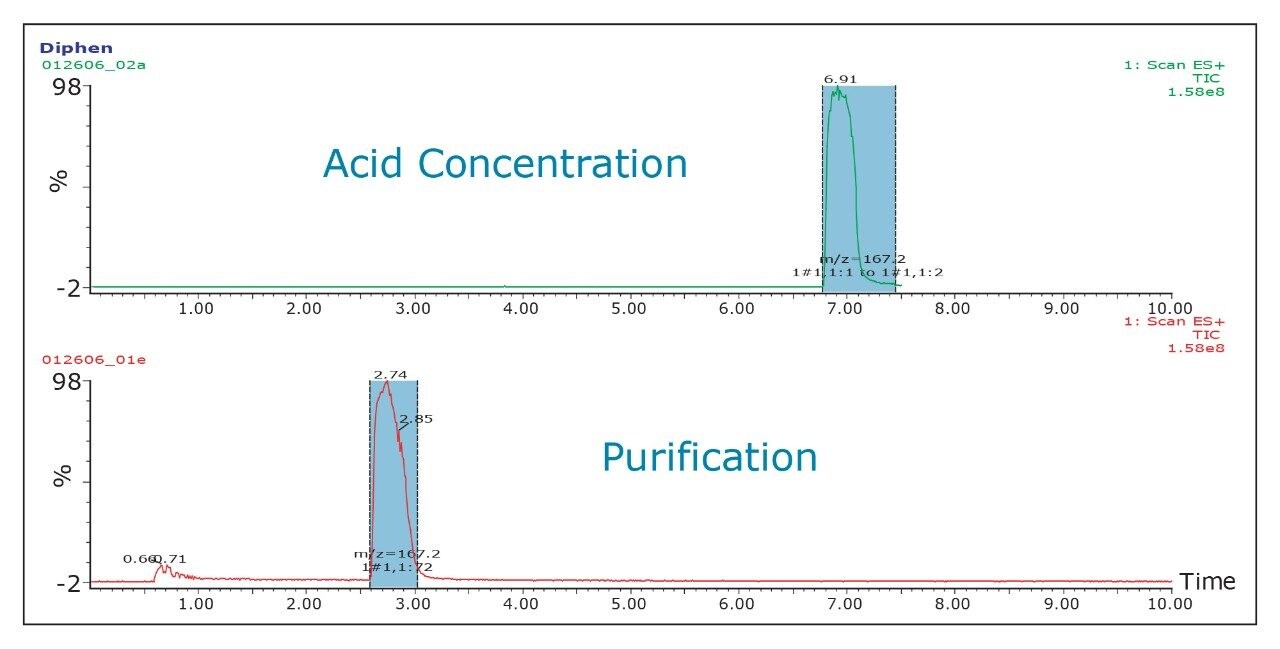
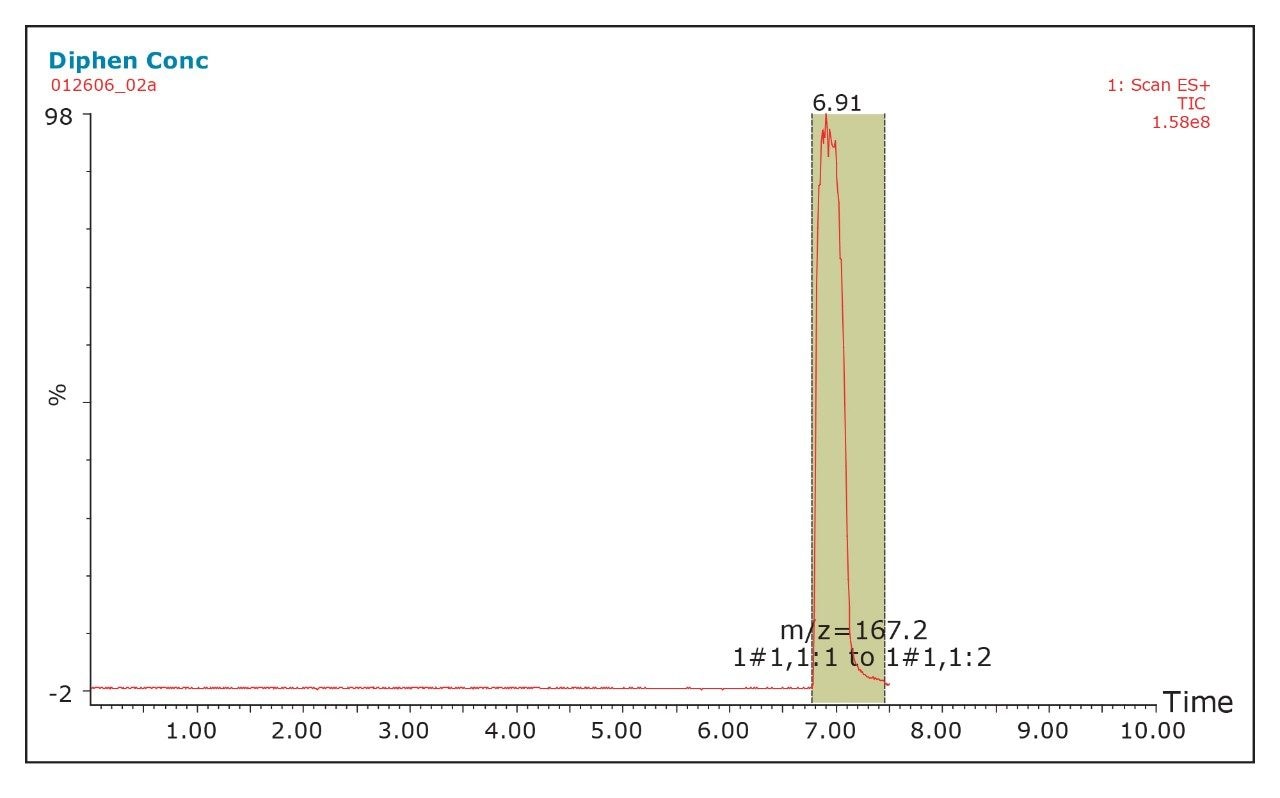
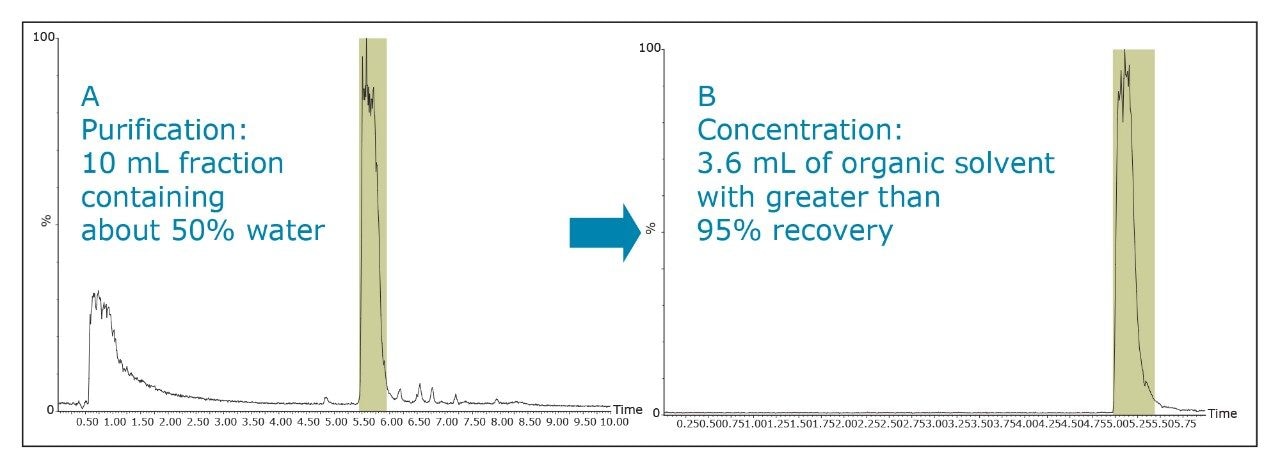
The pKa of the target compound should be considered when performing purification. The target compound should be neutral during the purification. This means that a basic compound should be run in a basic mobile phase and, conversely, an acidic target should be run in an acidic mobile phase.
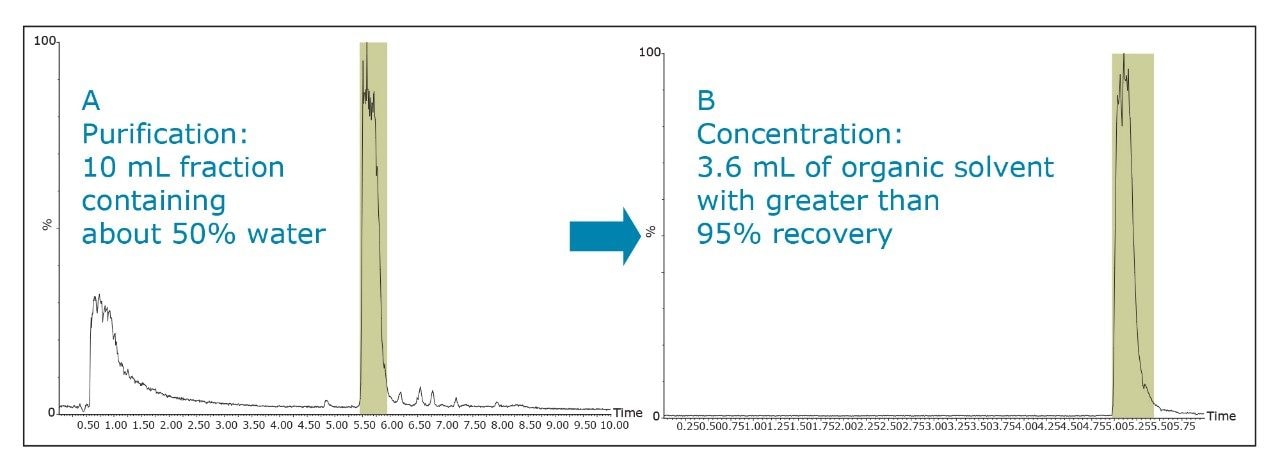
This will result in better loading and chromatography1 and will also ensure that the collected fraction is not ionized in solution. By being neutral, it is more likely to be successfully trapped during the concentration process.
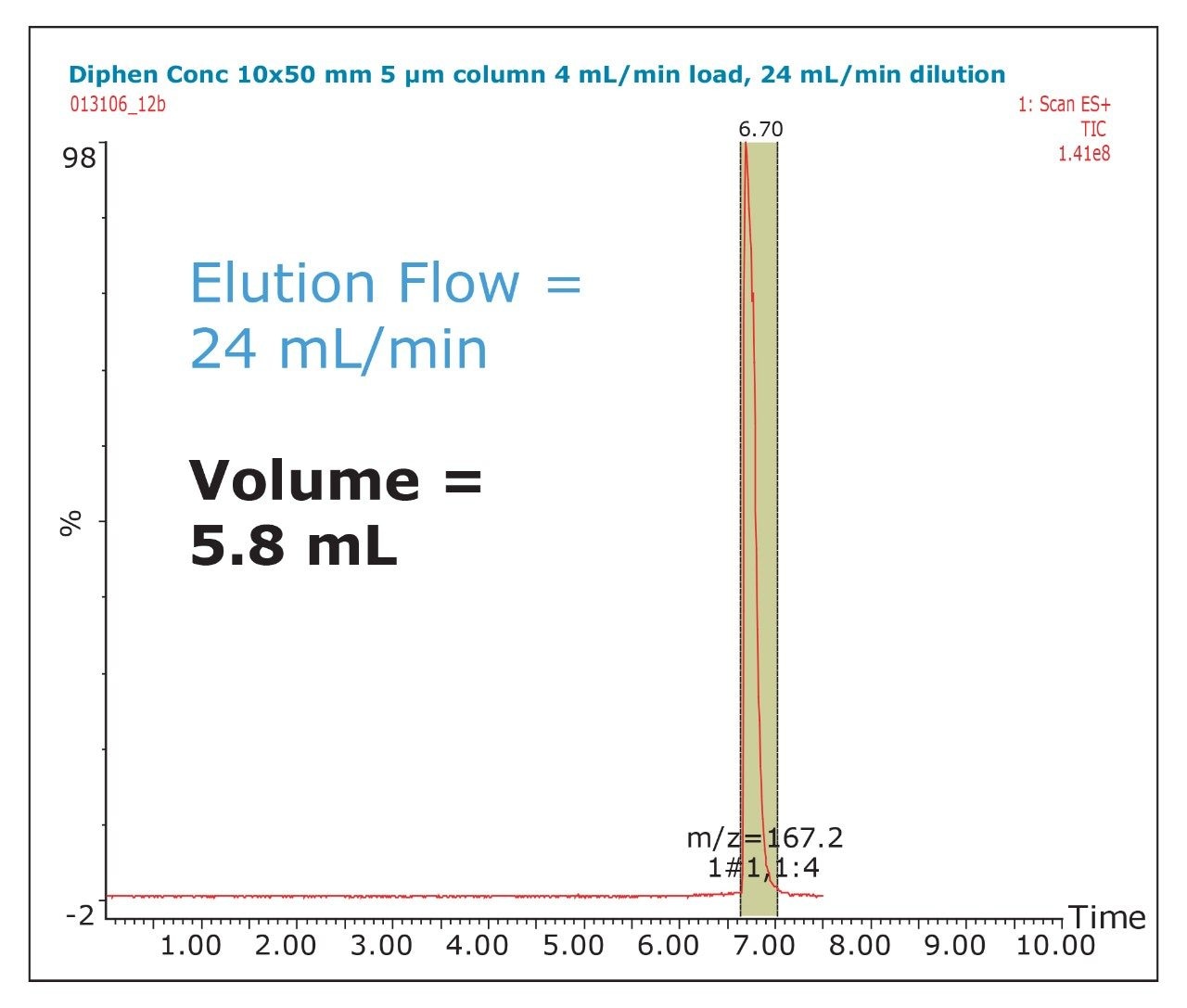
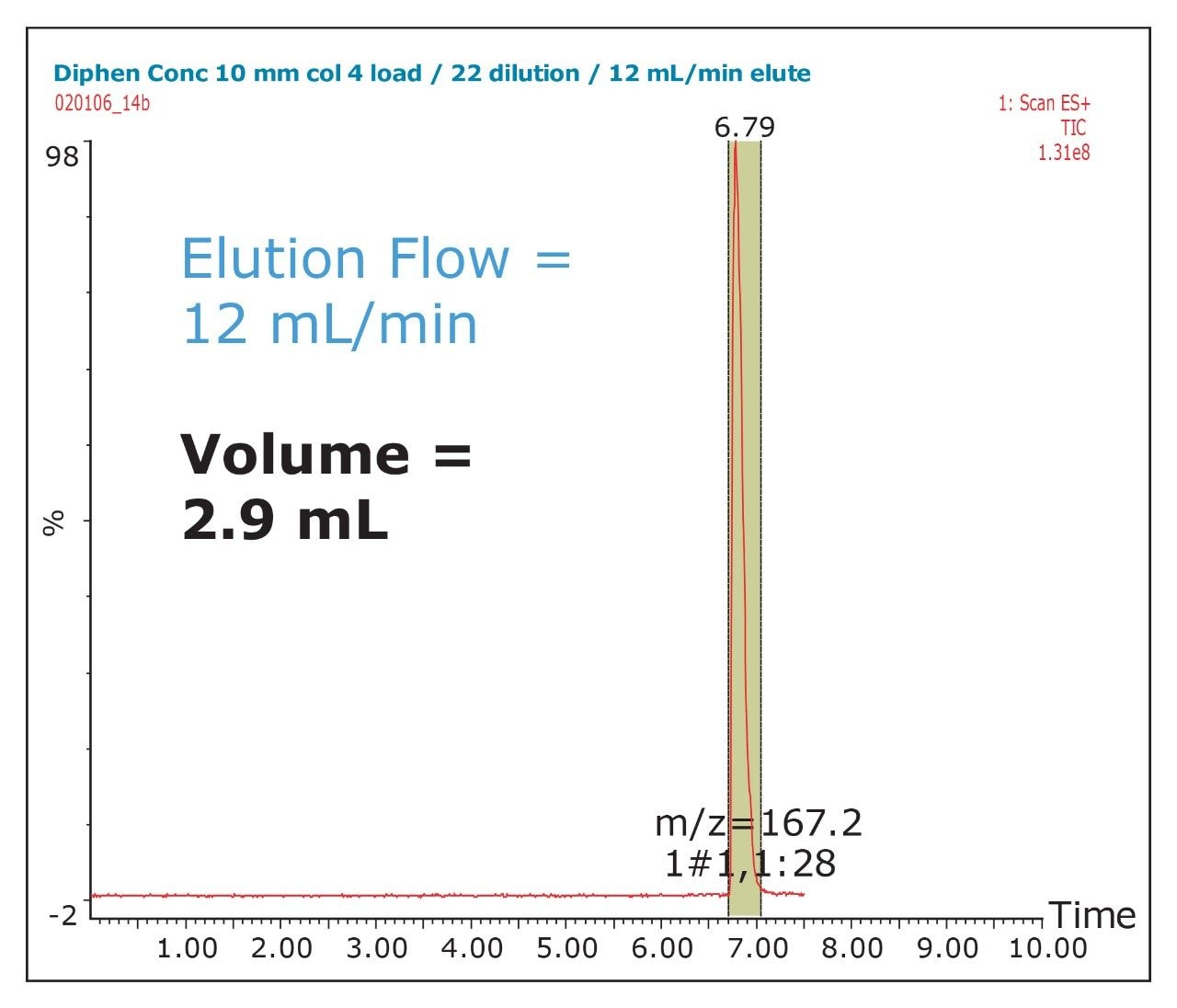
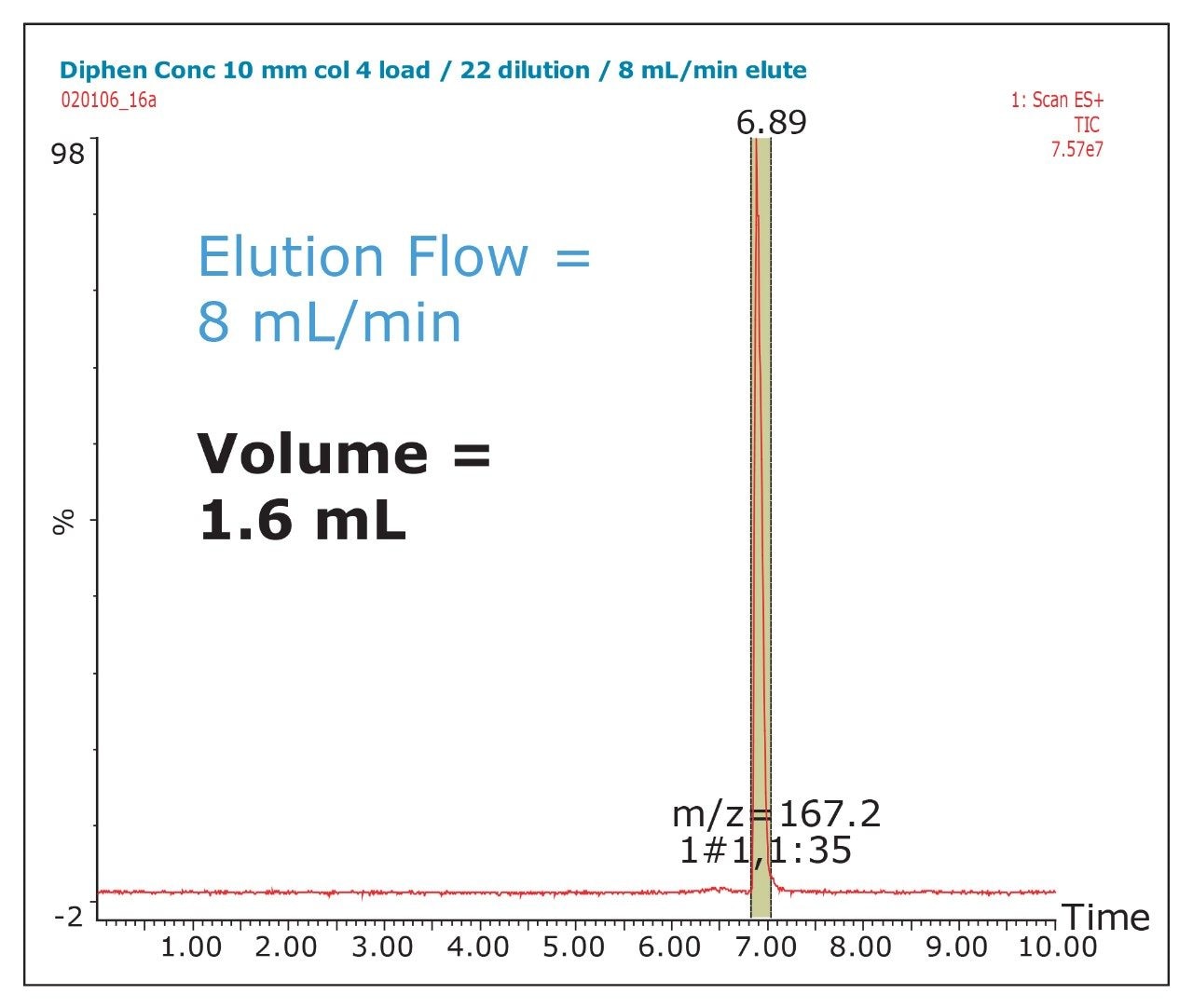
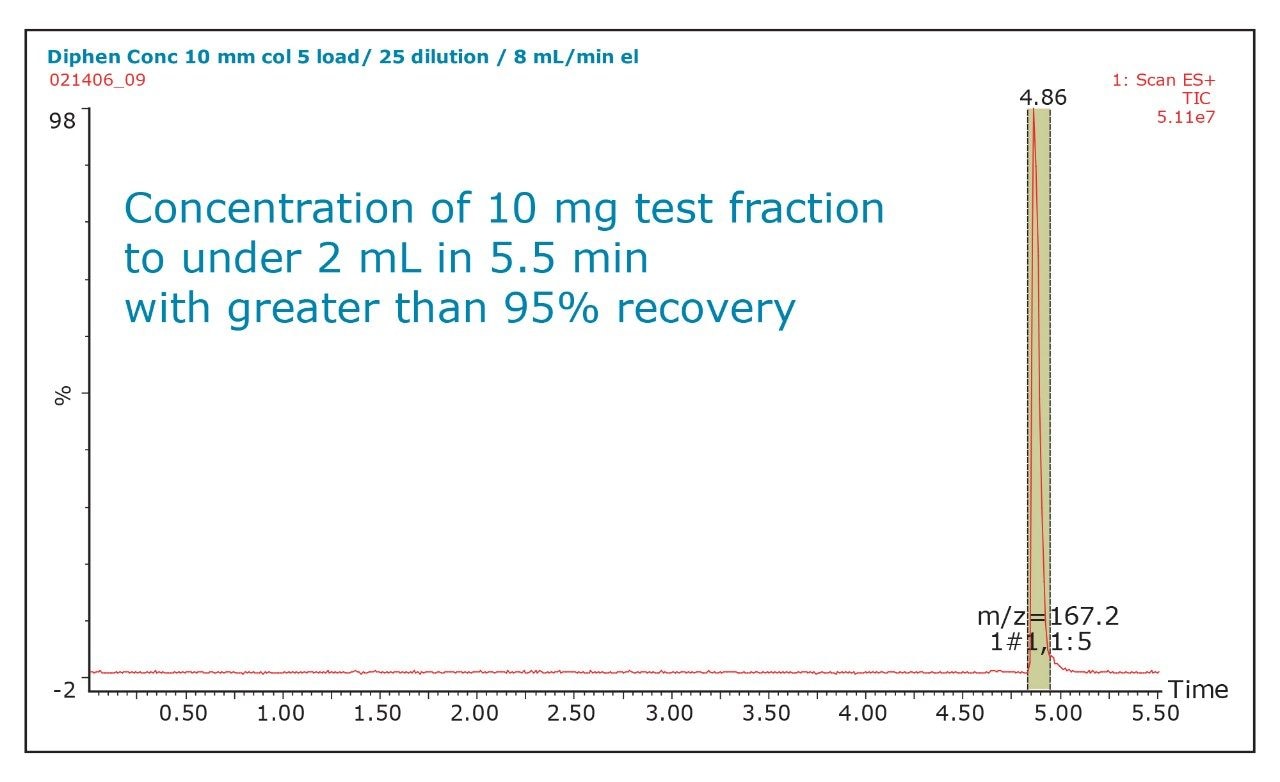
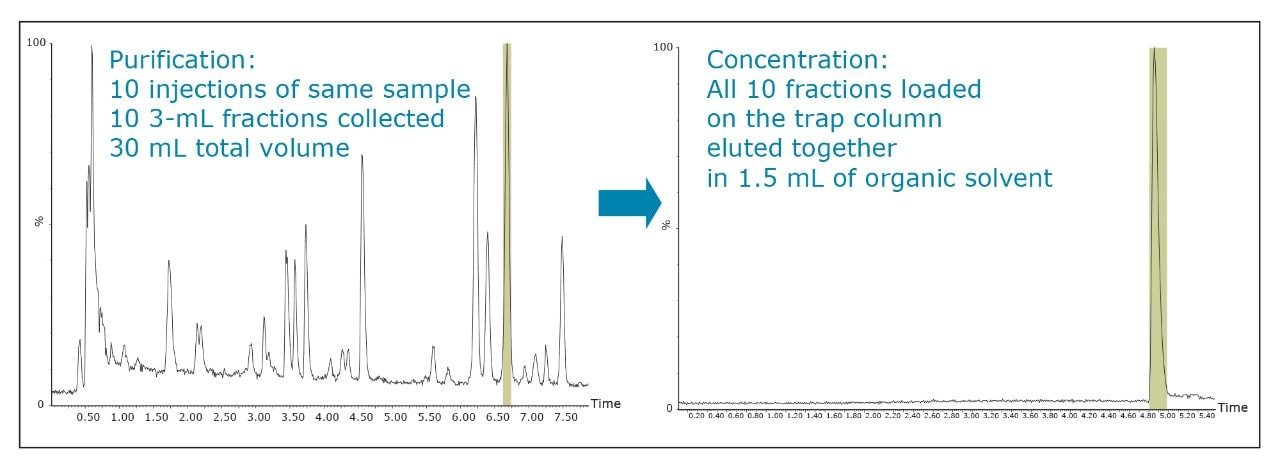
The amount of collected material, in both mass and volume, will dictate the required system configuration. The volume of the fraction will determine the size loop required. The mass of collected material will determine the column size. Both the loop and column size will determine the overall throughput of the system.
In the examples shown, all of the concentrated fractions were triggered by MS. However, this was done only for method development purposes. It is possible to collect these fractions by UV or just by time. When collecting by time, each tube has the same volume and organic concentration, so the time required for drying is constant. With typical fractionation, each tube can have a different volume and organic concentration, so the time required for drying is variable. This variability can lead to inefficiency, by either drying too long, or by stopping too early then checking multiple tubes to find that you need to restart for only a few of the tubes.