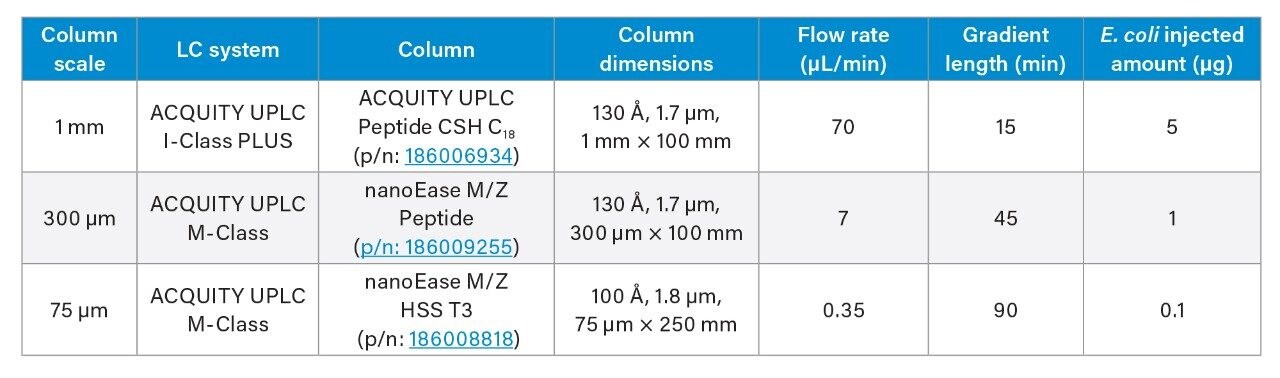
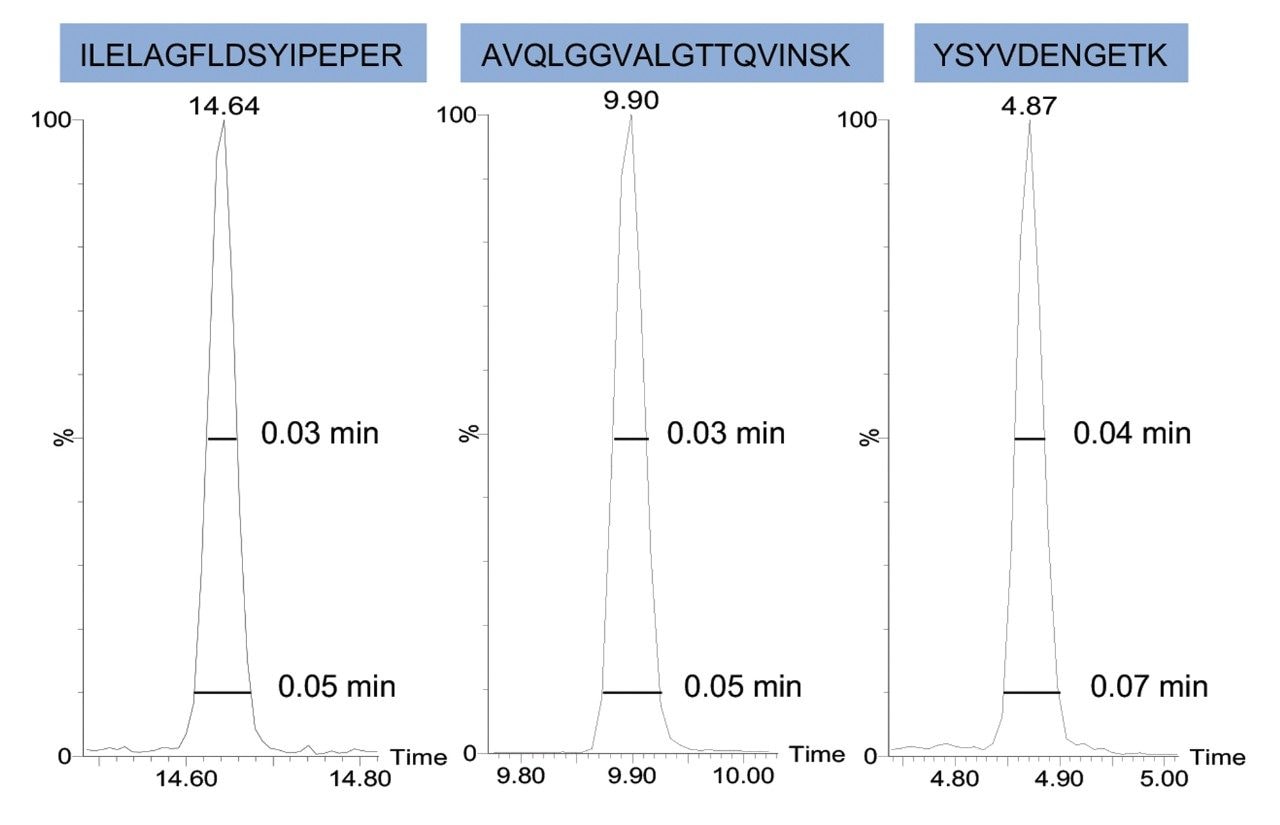
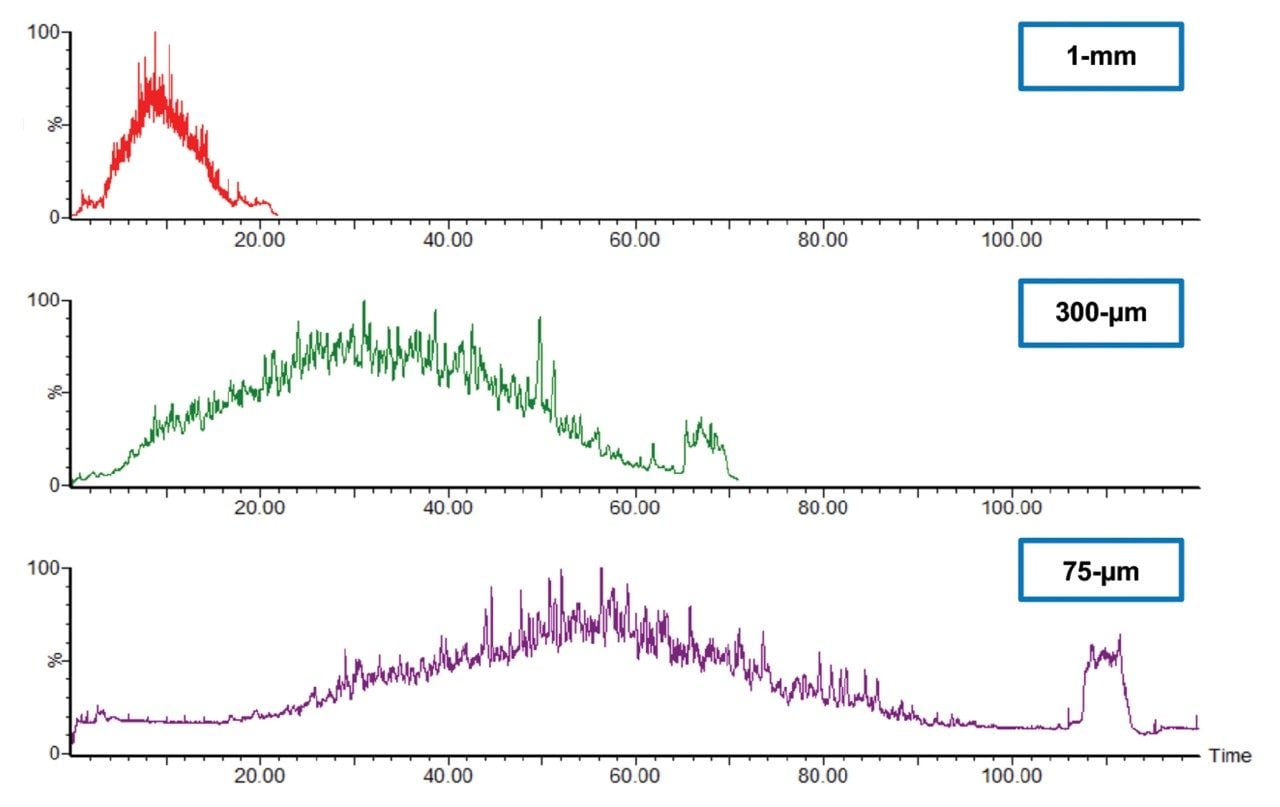
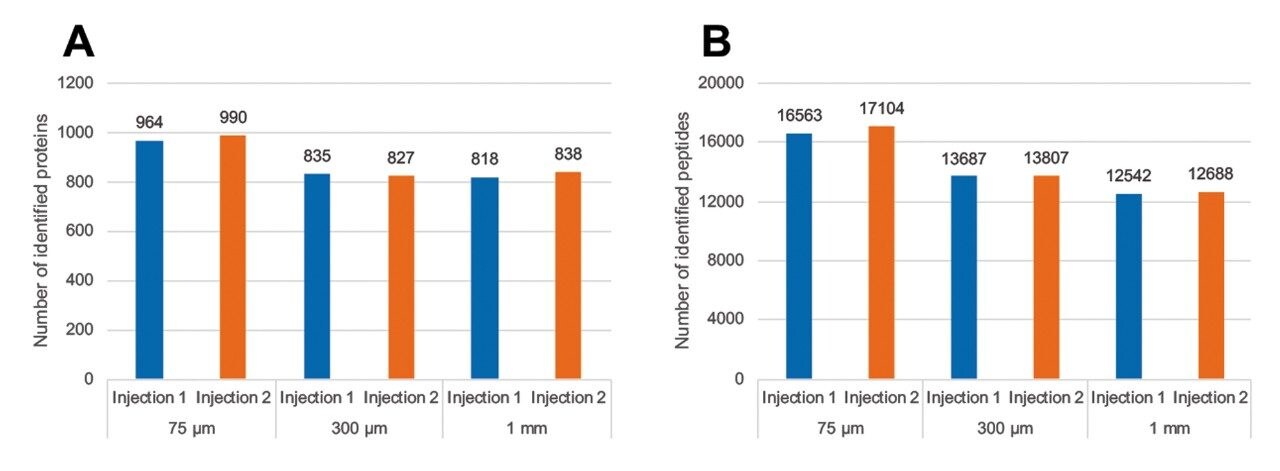
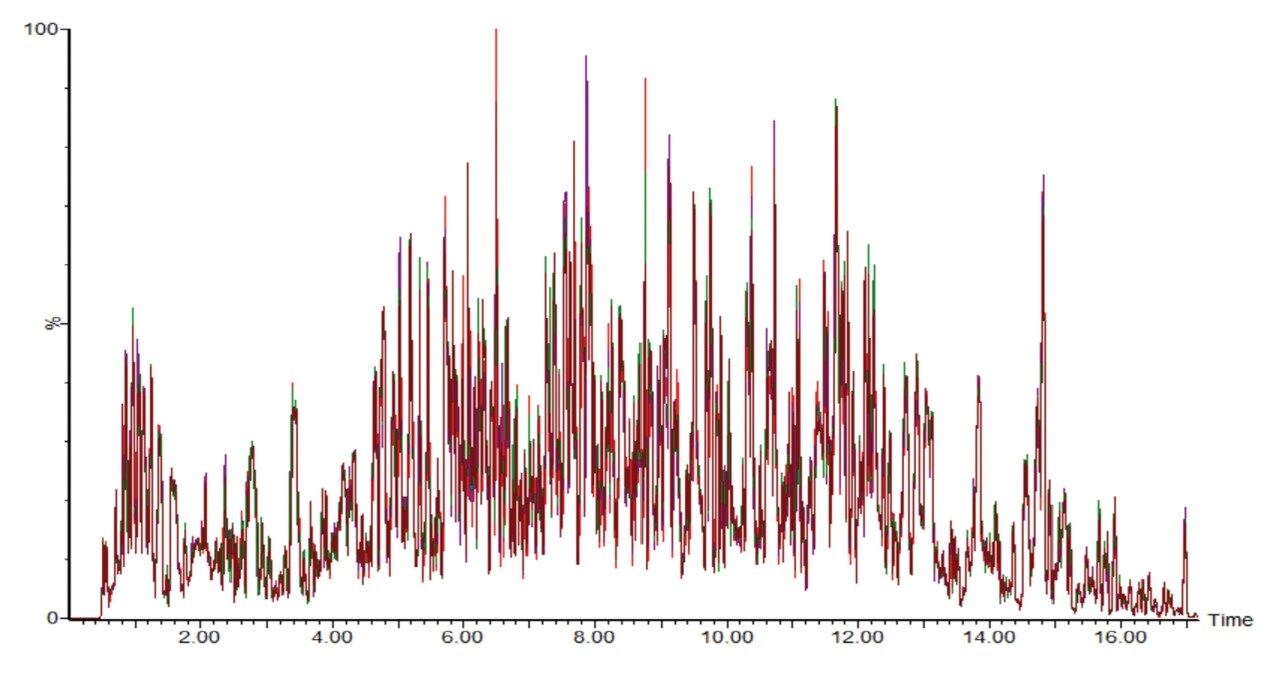
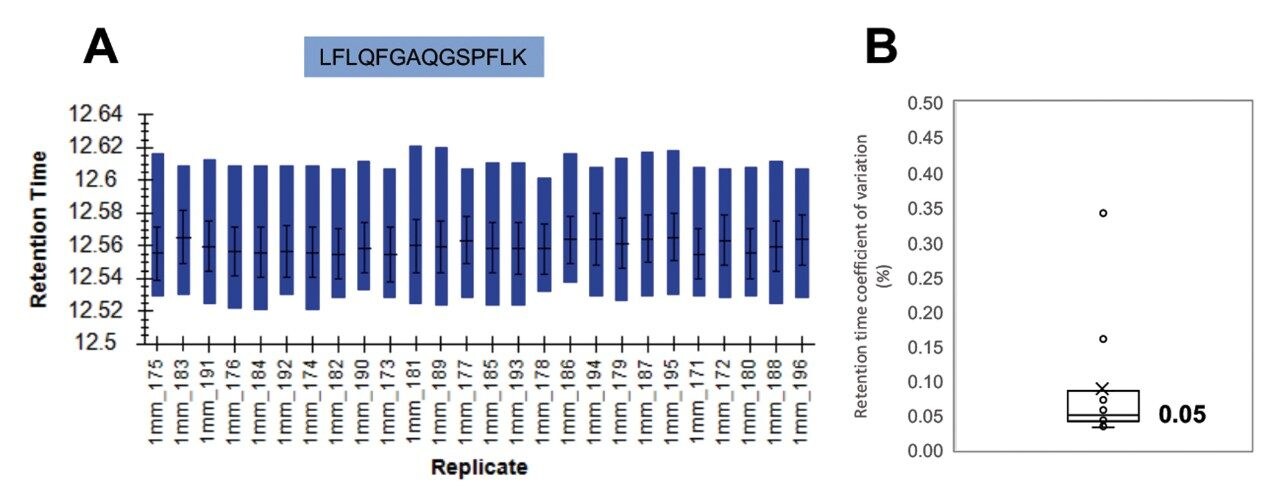
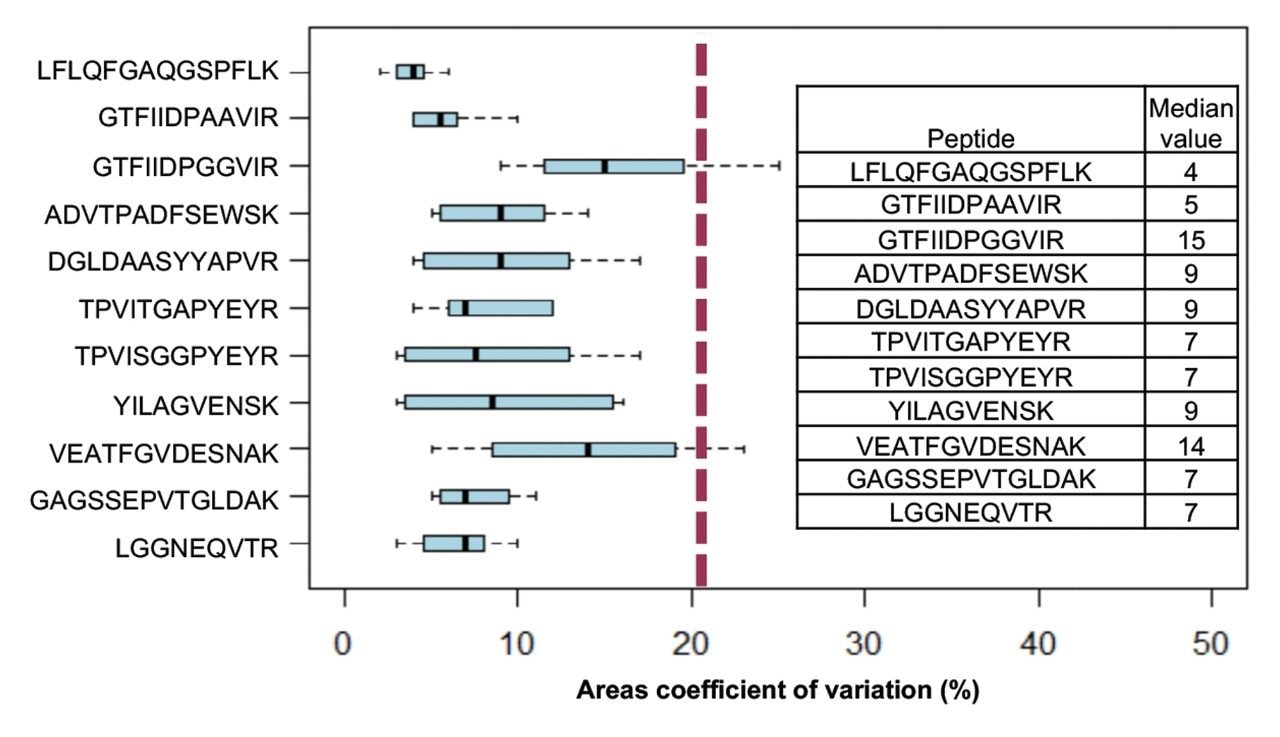
Today, there is an increased interest in clinical proteomics and its application for the analysis of large population cohorts. Traditional discovery proteomics typically uses nanoflow chromatography with 75 μm internal diameter columns using a flow rate between 0.2 and 0.5 μL/min, providing the high sensitivity and coverage for complex proteomic digests. However, long gradients and equilibration times are disadvantageous. For one sample, the total run time is typically between 60 and 180 min. Although the design of the LC has been largely improved for usability, for instance with finger tight fittings (i.e., Waters ZenFit Connectors), nanoscale chromatography remains challenging to use and troubleshoot, restricting its applicability for large cohort analysis. Recently, with improved mass spectrometry resolution and sensitivity, microflow scale chromatography using 300 μm column internal diameters with flow rates between 7 and 12 μL/min have been implemented to analyze large cohorts of digested human plasma samples.1,2,3 Here, we show the utility of 1 mm scale chromatography coupled to the SYNAPT XS Mass Spectrometer to analyze mid-complexity samples such as E. coli and human plasma digests in a reduced timeframe (i.e., 15-min gradient), while maintaining comparable protein/peptide identifications.