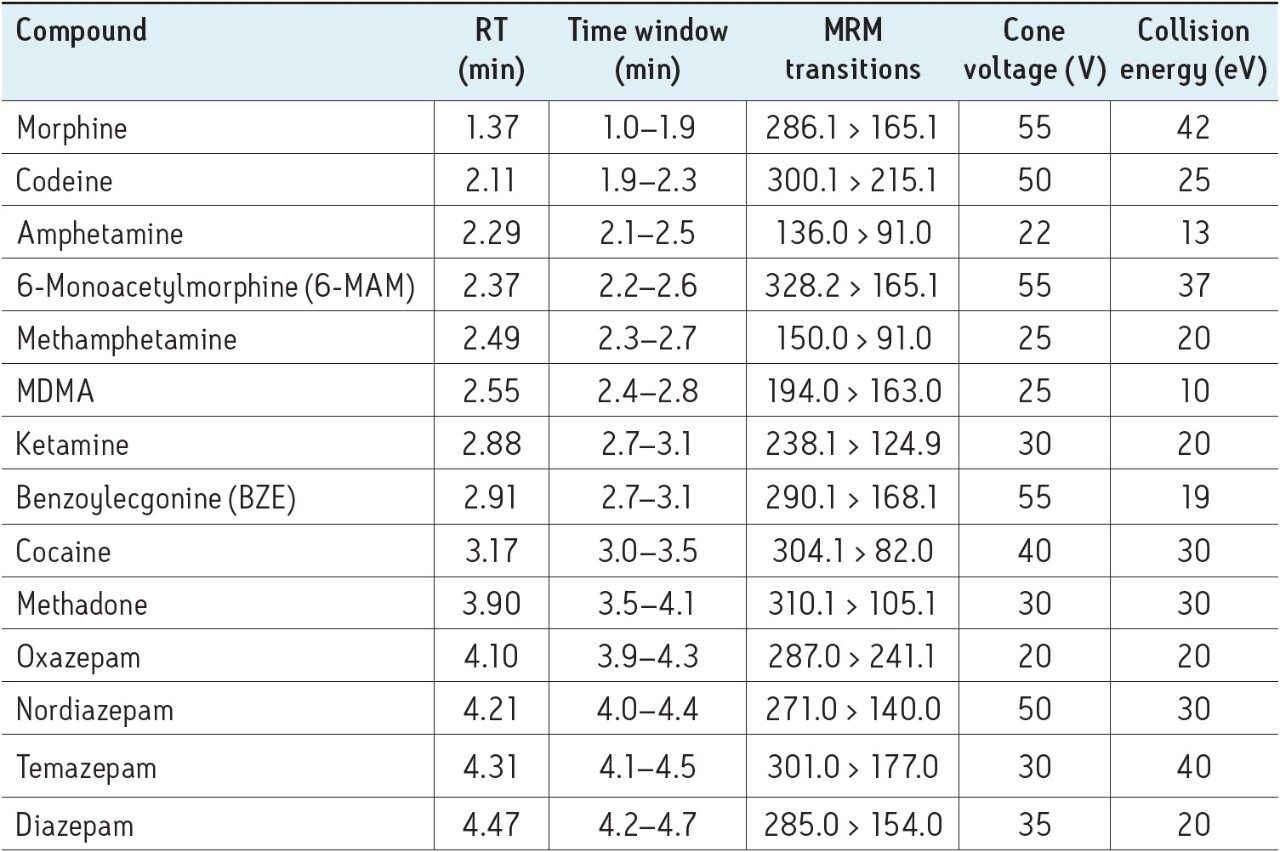
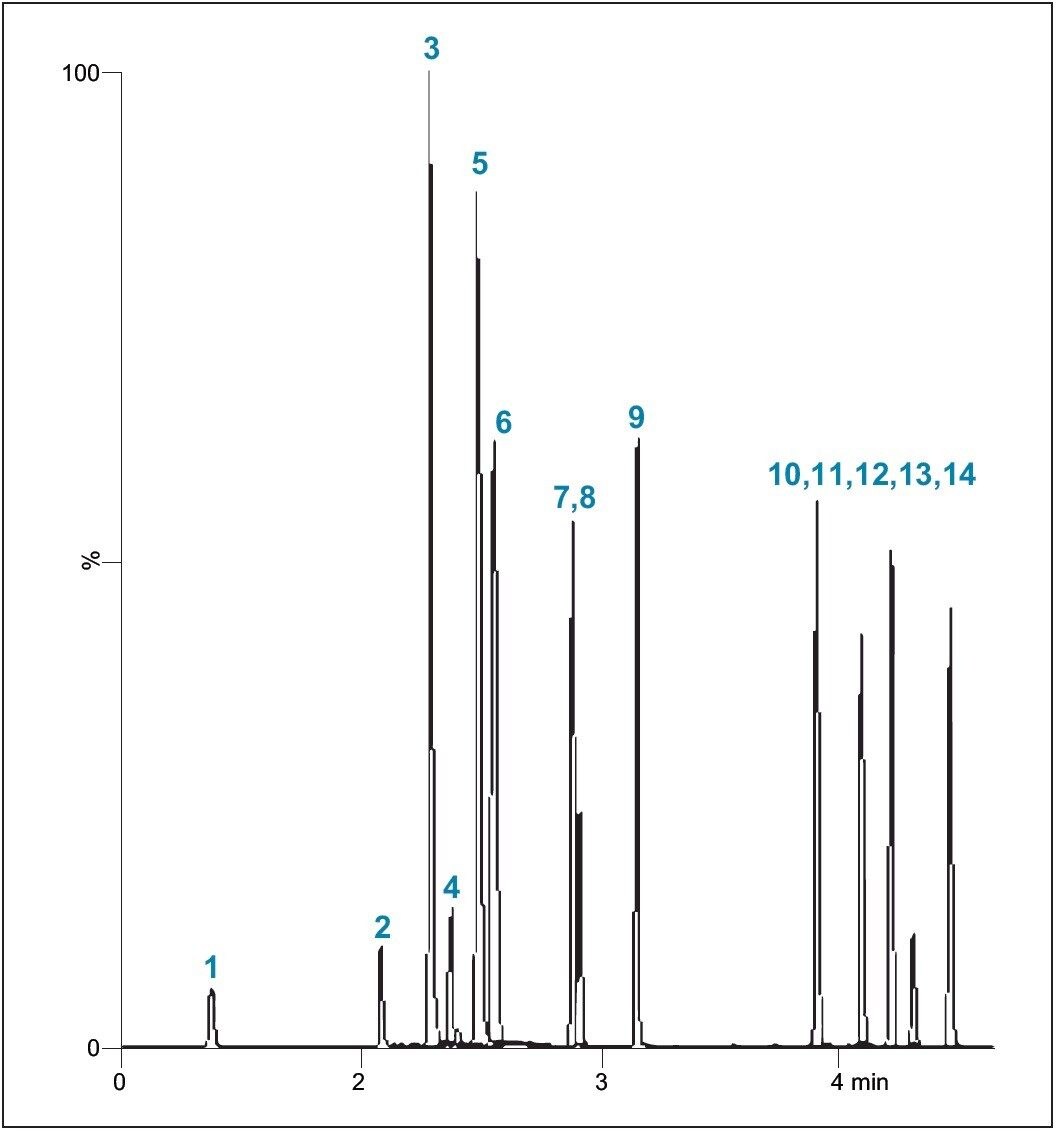
The acceptance criteria for a positive identification of analytes were: the retention time to be within 0.2 min of predicted and the quantifier/qualifier ion ratio to be within 20% of the predicted ratio, which was based on the average of the ratios across the entire calibrator range.
To investigate linearity for all analytes, spiked preserved oral fluid calibrators and quality control (QC) samples were prepared daily (the concentrations in neat oral fluid ranged from 0 ng/mL to 500 ng/mL) and analyzed on four different days. Peak areas for each MRM trace were generated automatically using the TargetLynx Application Manager and referenced to the appropriate ISTD peak area. Quantitative calibration curves were plotted using a 1/x weighting with a quadratic fit applied to all analytes. Interday coefficient of determination (assessed over four days) was >0.995 for each analyte.
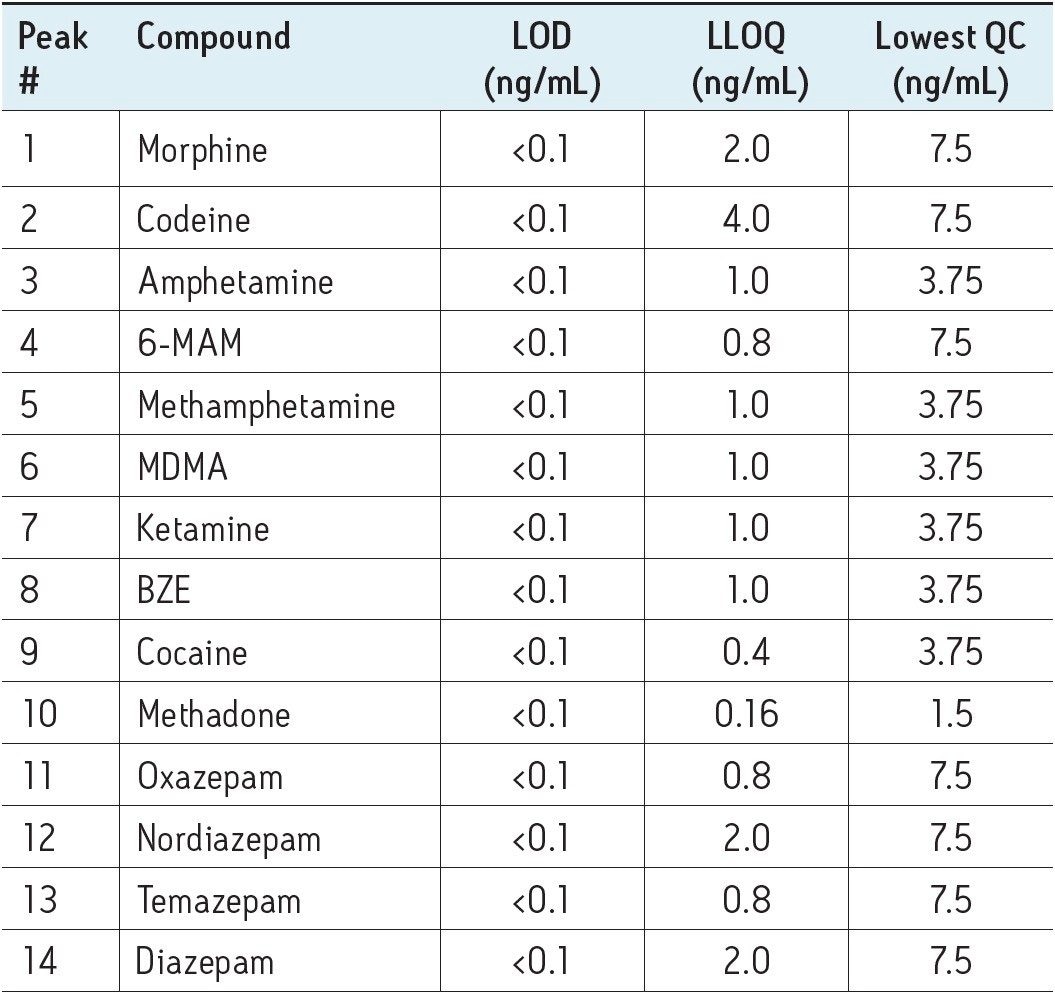
The limit of detection (LOD) was defined as the lowest concentration which gave a signal-to-noise ratio >10:1 (for both transitions) in spiked preserved oral fluid. The lower limit of quantitation (LLOQ) was defined as the lowest concentration which gave a signal to noise ratio >10:1 (for both transitions) and ion ratios within 20% of expected and the achieved concentration was within 20% of target with a %RSD of <20% in preserved oral fluid over the four day period. The LOD and LLOQ for each analyte are summarised in Table 2 along with the concentration of the lowest QC sample assayed. Extraction recovery and matrix effects for each analyte were investigated in six different sources of preserved oral fluid at three concentrations (5, 25, and 100 ng/mL), with the ISTDs at 12.5 ng/mL. The mean % recovery for each analyte was matched by that of the appropriate deuterated internal standard and was acceptable for this assay. The matrix effects were matched by the appropriate deuterated internal standard and were shown to be less than 25% for the majority of analytes with the %RSD less than 15% for all analytes.