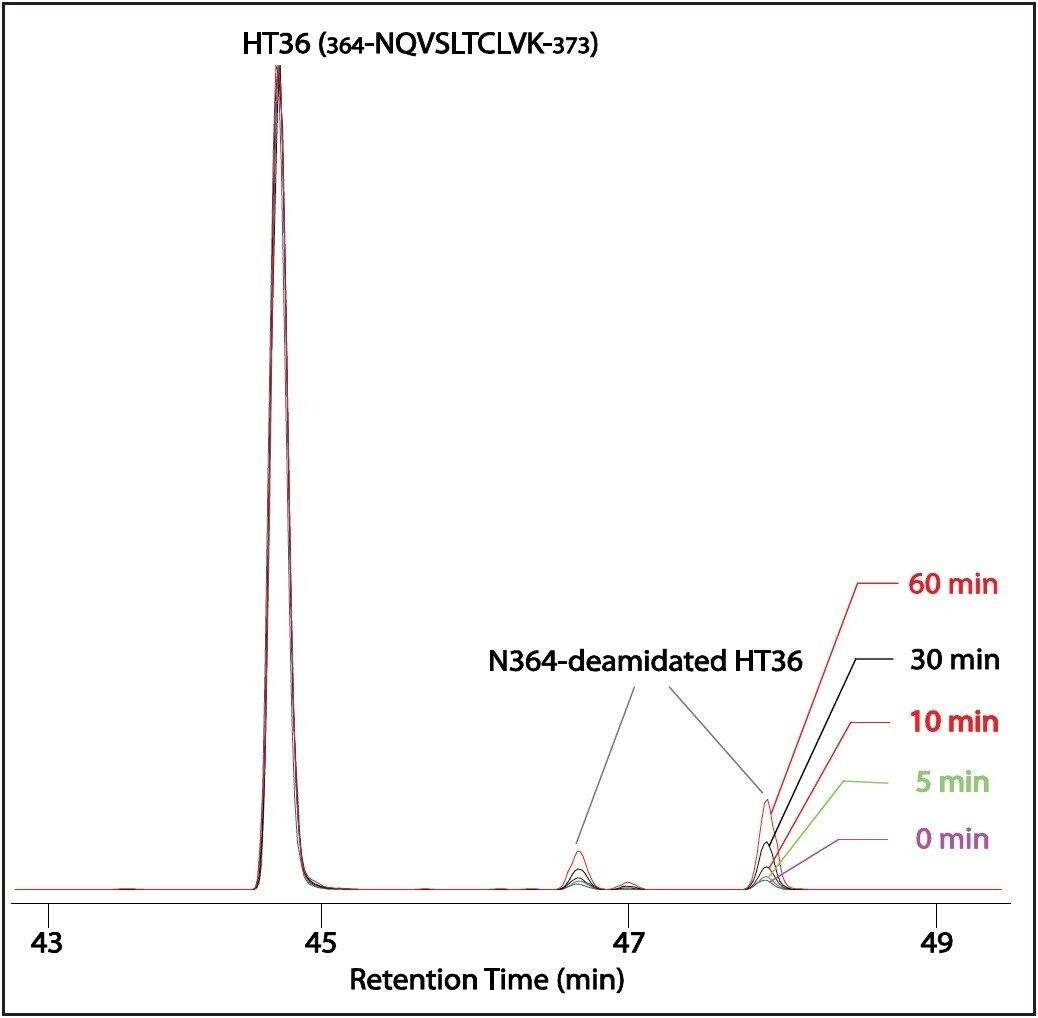
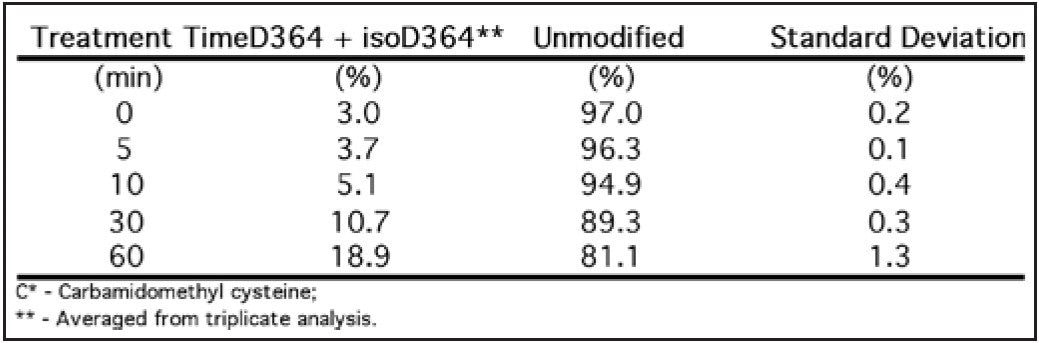
The detailed tryptic digestion procedure has been described previously.2 Briefly: at presence of 0.05% RapiGest SF, the mAb was digested with trypsin for 4 hours after reduction with dithiothereitol and alkylation with iodoacetamide. The pH of the resulting digest was then adjusted to ~ 9 with 1 M NH4OH. Aliquots of the alkalized digest were incubated at elevated temperature (60 °C) for 0, 5, 10, 30, and 60 min, respectively. Following incubation, each aliquot was diluted (to 1.5 pmol/μL) with 0.1% formic acid (FA) in 5% acetonitrile, and analyzed by LC-MSE peptide mapping experiment.
LC-MSE studies were performed using a Xevo QTof MS System coupled with an on-line ACQUITY UPLC System. It was configured with a 1.7-μm Peptide Separation Technology (PST) BEH300 C18 Column, 2.1 x 150 mm. Peptides were eluted with a 90-min gradient (1 to 40% acetonitrile in 0.1% FA) at a flow rate of 0.2 mL/min and 60 °C column temperature.
MSE data were acquired at 1 Hz in ESI positive ion mode, with collision cell energy alternating between low energy (4 V) to collect peptide precursor (MS) data and elevated energy (ramping from 15 to 45 V) to obtain peptide fragmentation (MSE) data. A capillary voltage of 3.0 kV, source temperature of 105 °C, cone voltage of 25 V, and cone gas flow of 10 L/h were maintained during the analyses. Sampling of the LockSpray channel (100 fM GFP in 50:50 acetonitrile/water containing 0.1% FA) was performed every 1 min to ensure mass accuracy. The analyses were completed in triplicate.
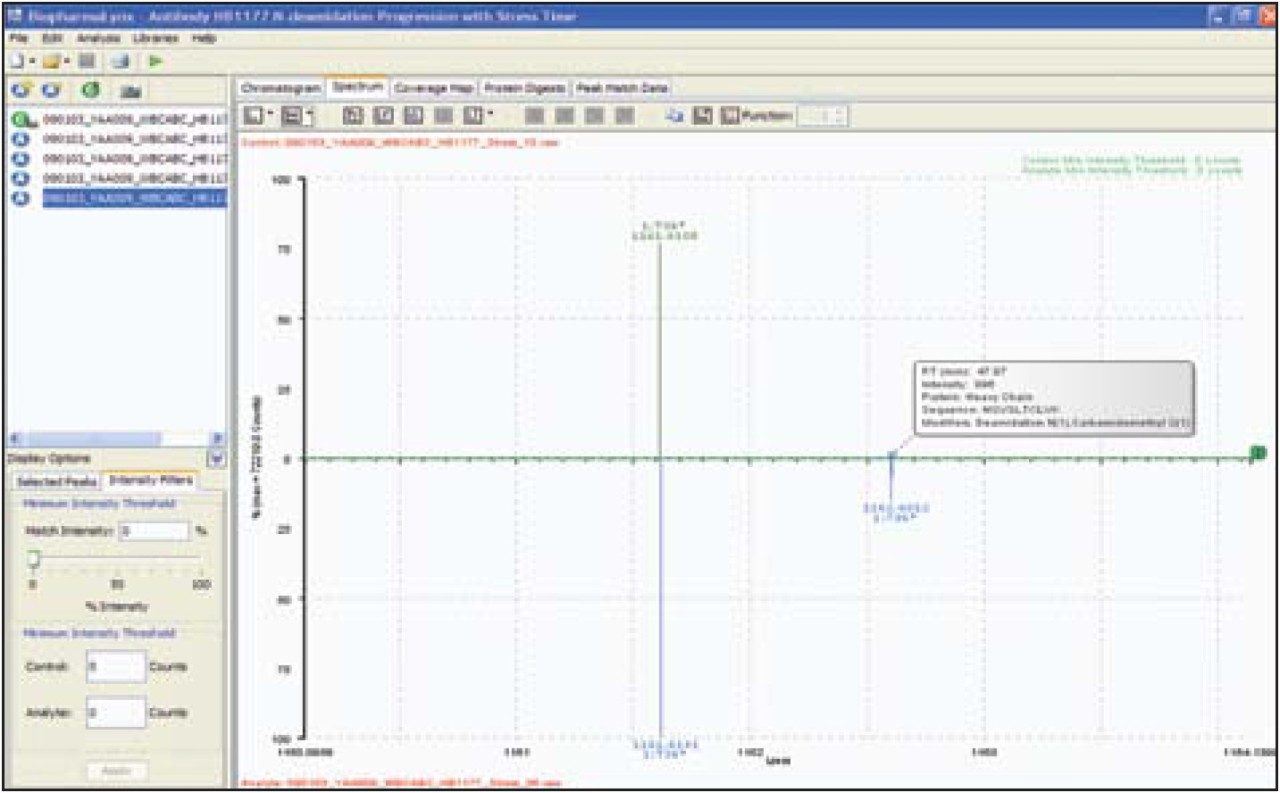
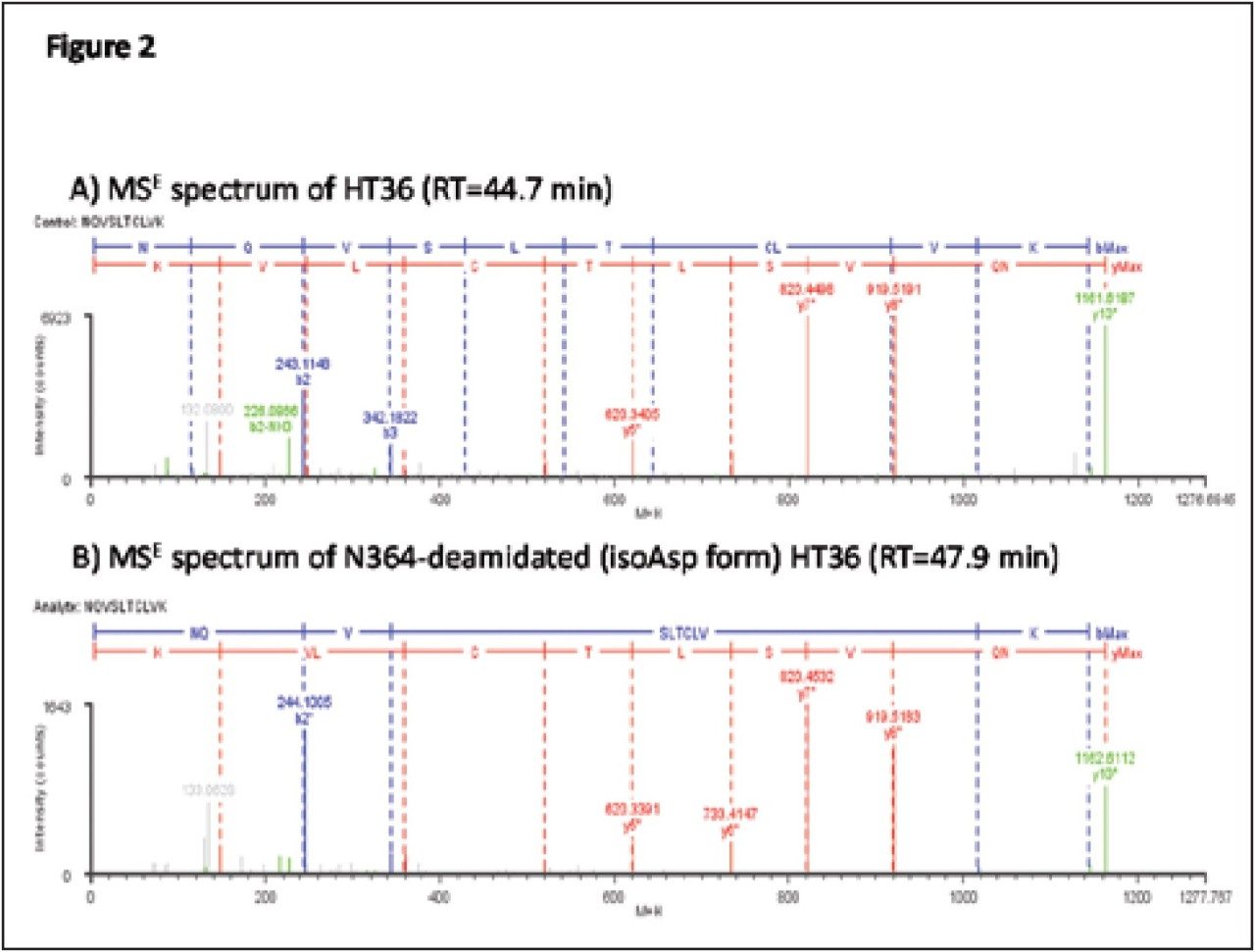
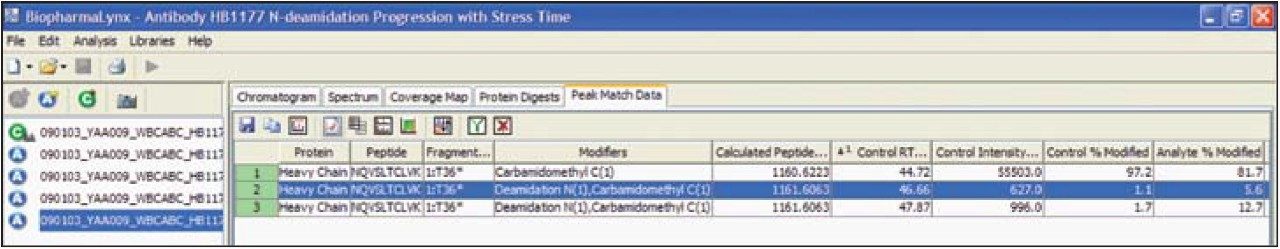
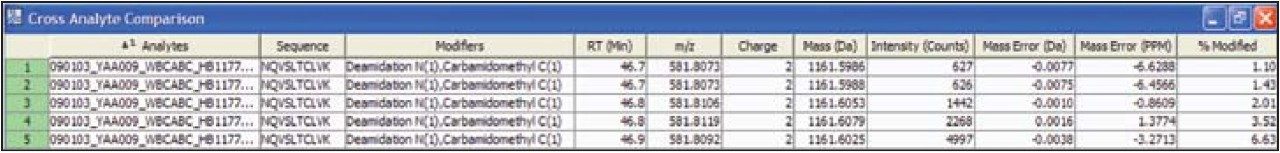
Peptide mapping data were batch-processed by BiopharmaLynx 1.2, an application manager for MassLynx Software, using traditional tryptic cleavage rules and setting cysteine carbamidomethylation as a fixed modification and N-deamidation as a variable modification. Additional BiopharmaLynx method parameter settings were detailed in a previous publication.3 Identifications of N-deamidated peptides were confirmed using the MSE fragmentation data that also enabled the peptide deamidation sites to be determined. The concentration of deamidations was calculated using reliably measured MS signal intensities.