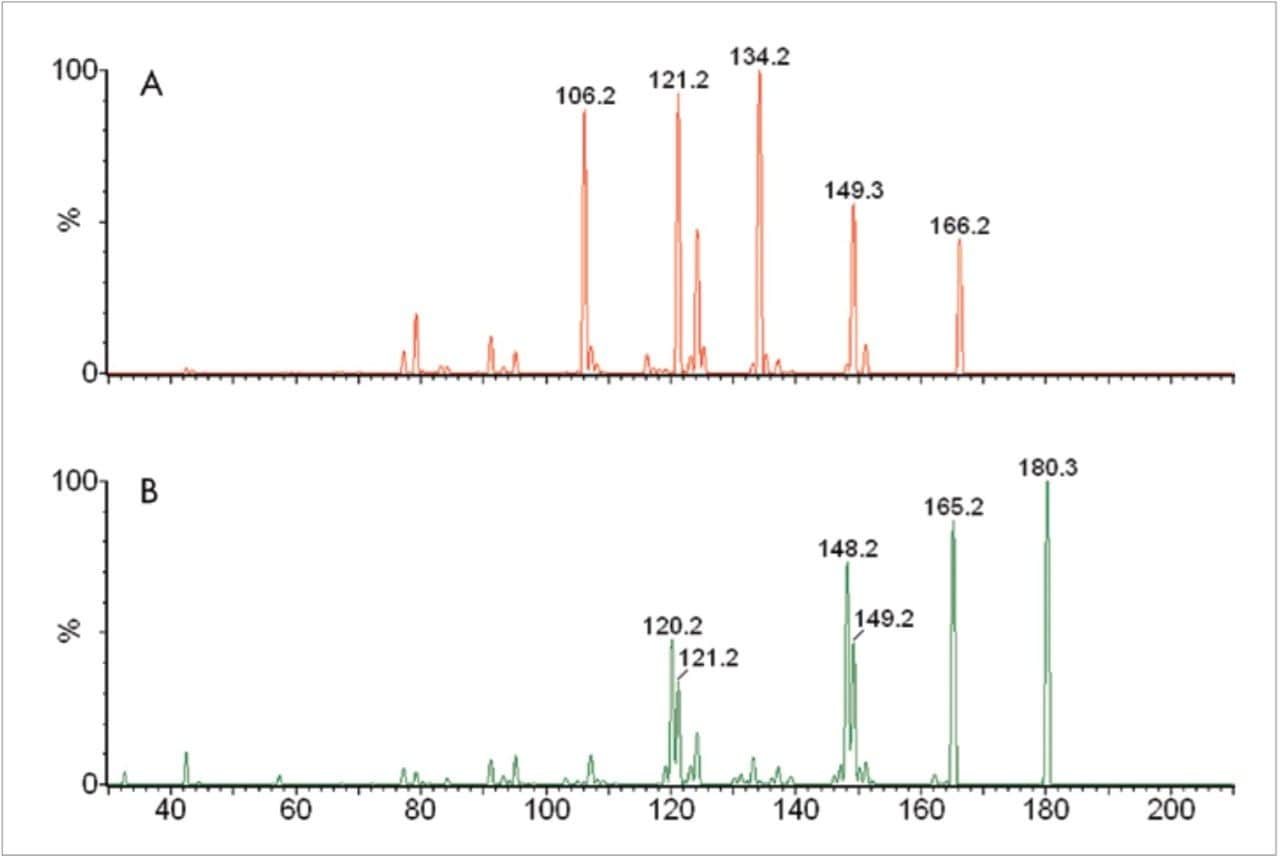
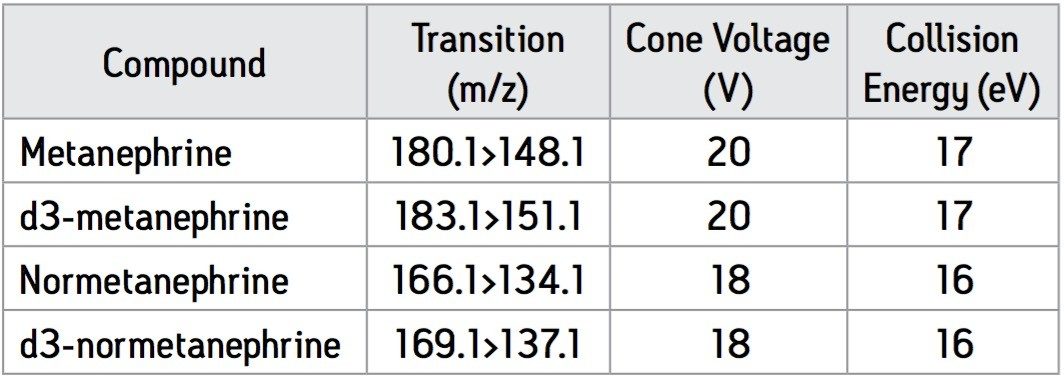
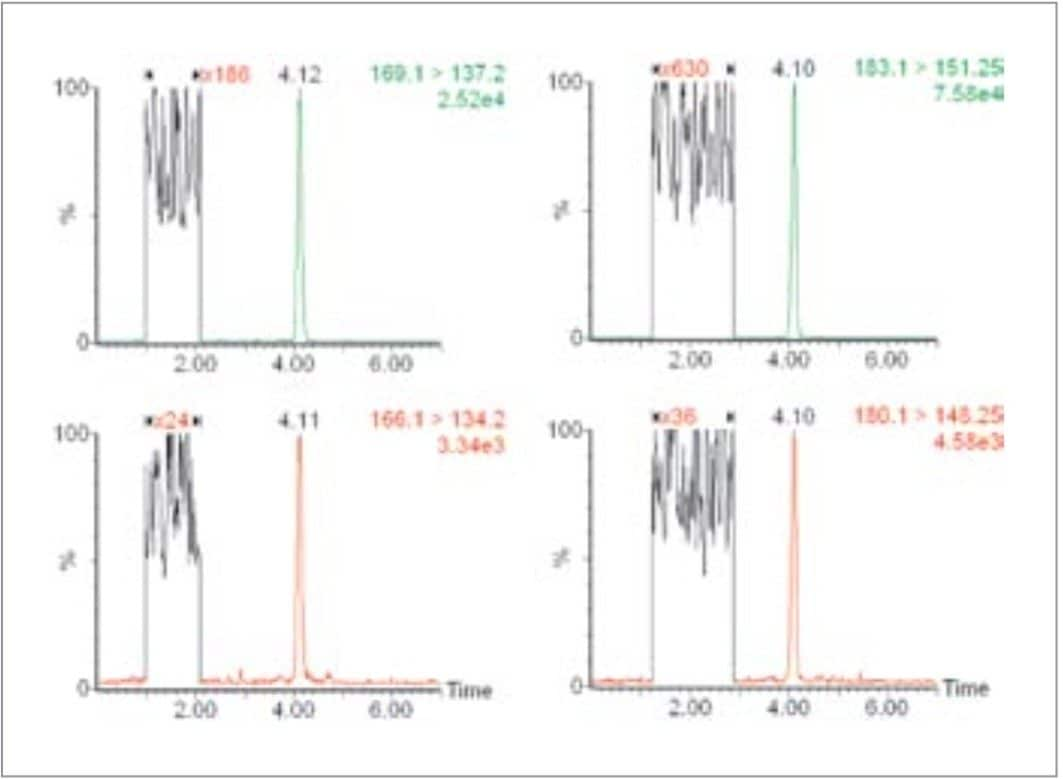
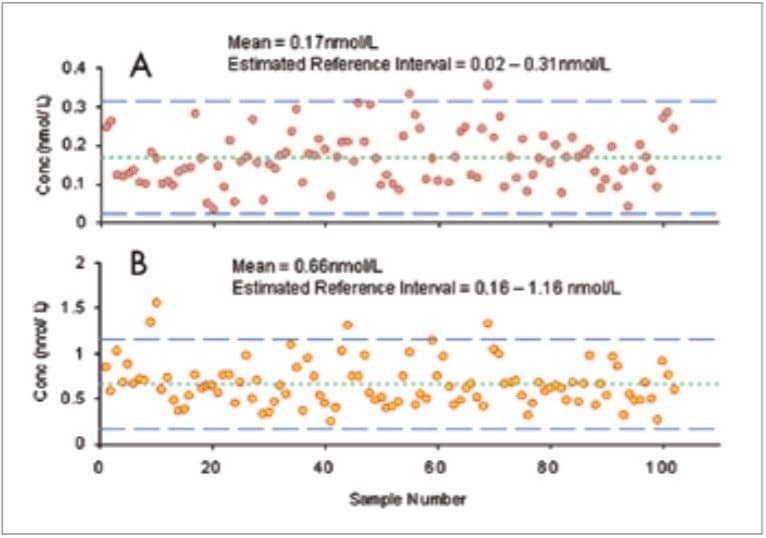
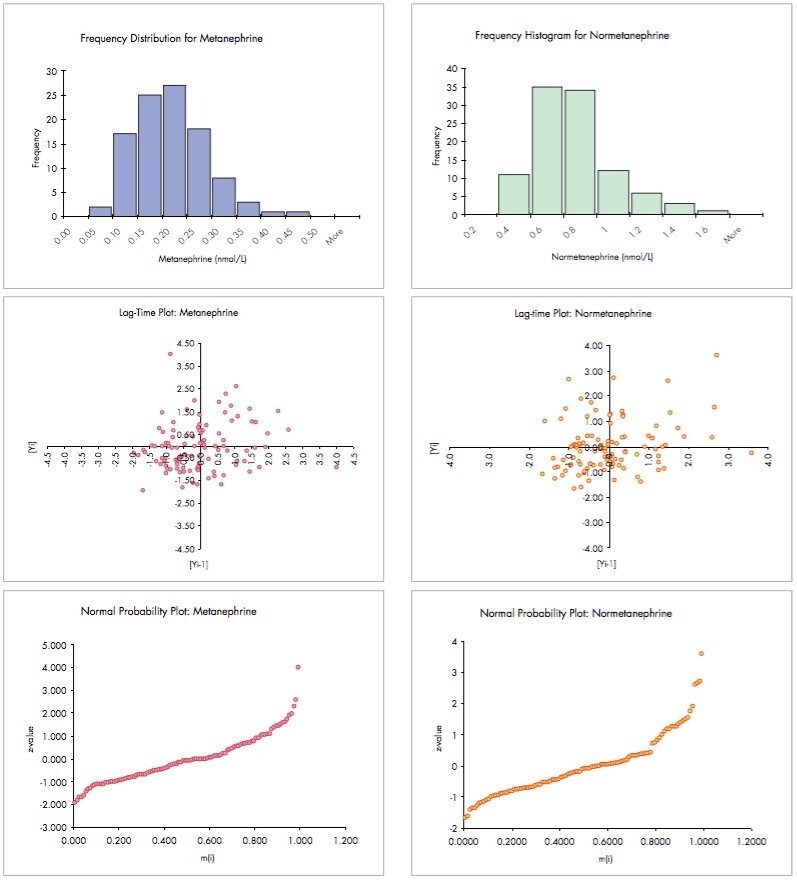
The frequency histograms for both M and NM show the expected non-gaussian distribution of levels in this group of patients. This phenomenon is more clearly demonstrated in the normal probability plots for these analytes – normal, gaussian distributions would have symmetrical frequency histograms and approximately linear normal probability plots.
A lag-time plot is intended to verify whether a data set or time series is random or not. If data appear to have no detectable pattern in a lag-time plot, as shown above, they may be regarded as being random. Detectable patterns in lag-time plots, such as rings, may indicate sinusoidal variation or a drifting of results during an analysis that warants further investigation.
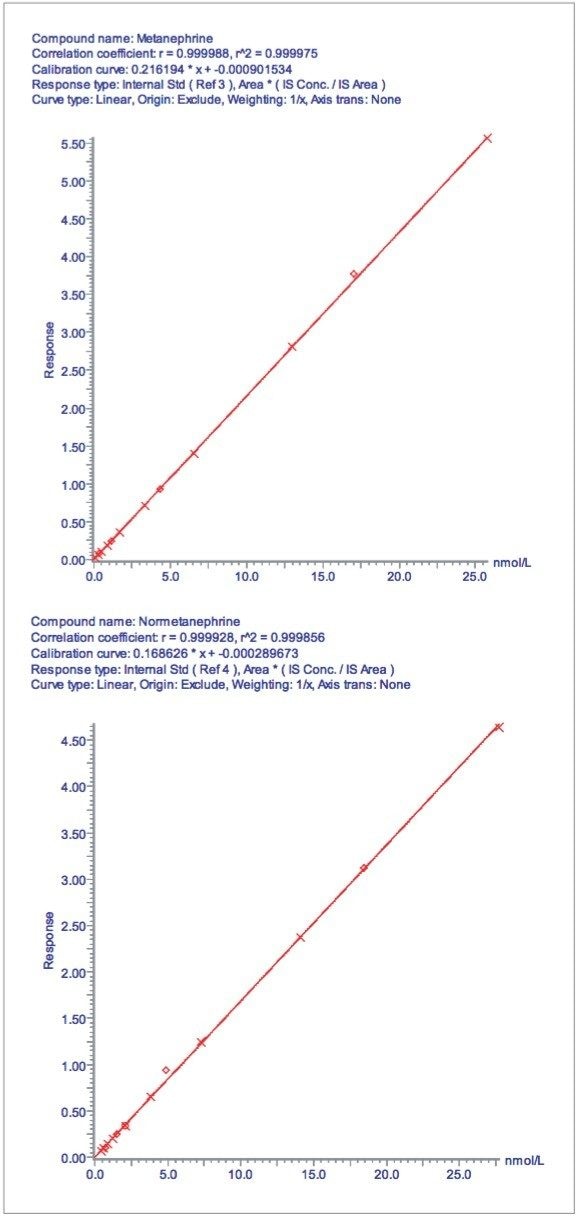
The use of on-line solid phase extraction technology coupled to LC-MS/MS has been shown to provide a plasma free metanephrines assay with improved sensitivity, selectivity and vastly reduced sample handling. Simple dilution of plasma samples with water containing deuterated internal standards followed by centrifugation can now be used to replace tedious off-line extraction methods.
A highly-selective extraction process is achieved using weak cation exchange (WCX) media. Traditionally, strong bases are extracted using strong cation exchange (SCX) media where the base must be eluted via neutralization. In the case of quarternary amines, this is often not possible and, more commonly, the stabilities of the basic analytes are compromised. Using Oasis WCX cartridge, strong bases bind to the carboxyl ion-exchanger at pH >5 permitting the cartridge to be washed with water and 100% acetonitrile without elution of the analytes of interest. Elution of the cartridge is then carried using the acidic mobile phase used in the chromatographic method and passed directly to the analytical column. The ability to wash the Oasis WCX extraction cartridge with 100% organic solvent whilst maintaining retention of the analytes of interest allows known ion-suppressing and non-polar species such as phospholipids to be removed prior to chromatographic analysis. This leads to a significant reduction in the likelihood of interferences and matrixdependent effects upon the assay.
The use of HILIC chemistry for the analysis of polar bases provides LC-MS/MS assays with higher sensitivities than traditional reversed-phase methods when using electrospray ionisation. The analytes of interest elute in high concentrations (circa 75%) of organic solvent where the desolvation process is more efficient. Using reverse-phase stationary phases, the metanephrines exhibit exceptionally poor retention and require the use of mobile phases of near 100% aqueous content or MS-unfriendly buffers.
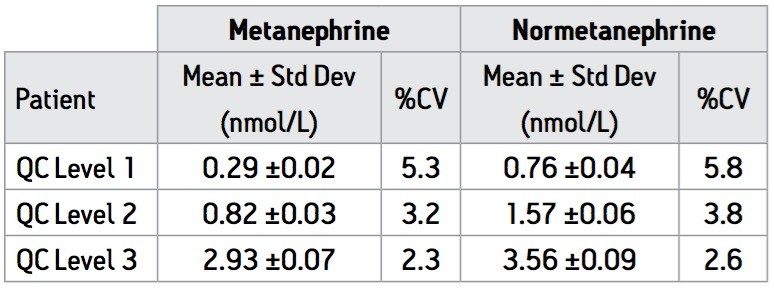
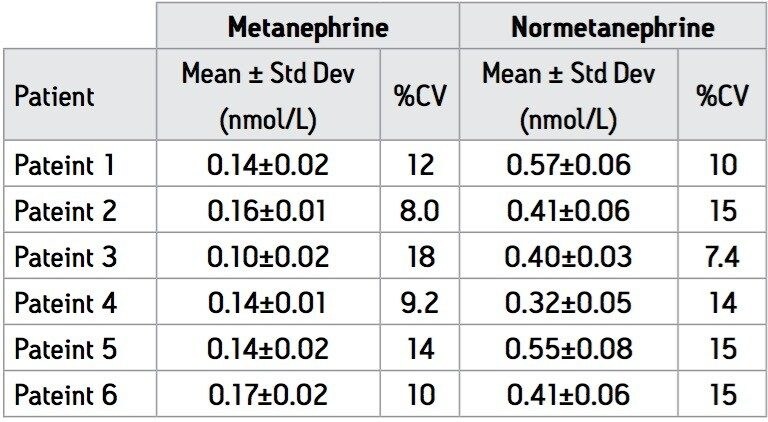
As a preliminary indication of the validity of the assay, the M and NM levels in the small group of patient samples (n=102) was used to calculate estimated reference intervals (Figure 4). These were found to be in close agreement with those in a previous study10 that suggests reference intervals of 0.05–0.47 nmol/L and 0.12–1.2 nmol/L for M and NM, respectively. Statistical treatment of these data has shown the data set to be random with a nongaussian distribution which is consistent with the expected results.
It should be noted that specimen collection strategies may have important consequences on the M and NM levels, particularly the position of the patient when blood samples are obtained. Since this information is not known for the samples used in this study, a more controlled study should be undertaken using a larger group of patients to provide reference intervals with greater credibility.
During the course of this study, heparinised and EDTA plasma samples were obtained from one patient to investigate the influence of two common anti-coagulants on M and NM results. No appreciable difference in the results was seen between the two samples when analyzed using this method. It is anticipated that the use of heparinised plasma sample may lead to sample stability issues owing to the formation of micro-clots in collected samples. Owing to the greater selectivity offered by XLC-MS/MS, the influence of sample matrix on the results should be reduced significantly.