During the manufacturing and packaging of active pharmaceutical ingredients (APIs), the removal of drug residues from the equipment is usually performed by a series of cleaning procedures. It is imperative that the production equipment used in this process be properly cleaned in order to avoid cross-contamination of drug products.1-3
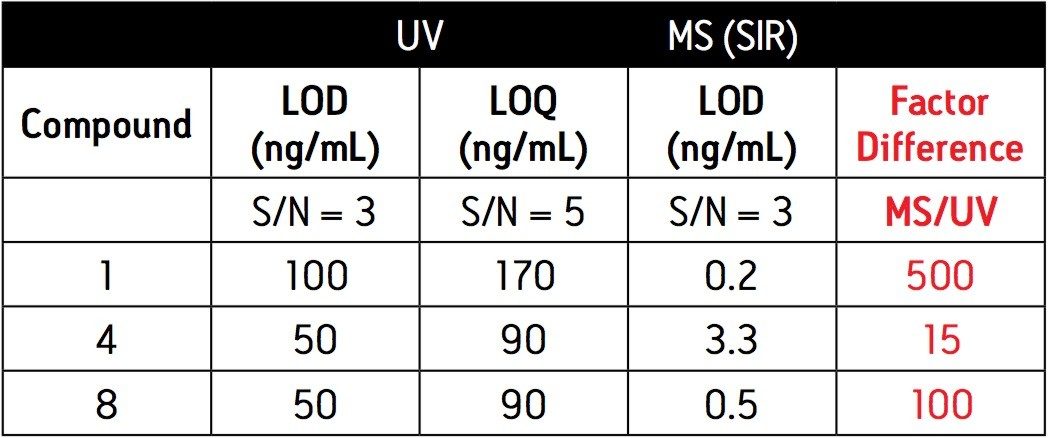
The safety acceptance criteria for API residues vary with drug substance. More potent compounds will require a lower acceptance limit. In general, most processes aim to have a lower safety limit in the 10 ppb–1 ppm range (10 ng/mL–1 μg/mL). In order to achieve these limits, sensitive analytical techniques are required.
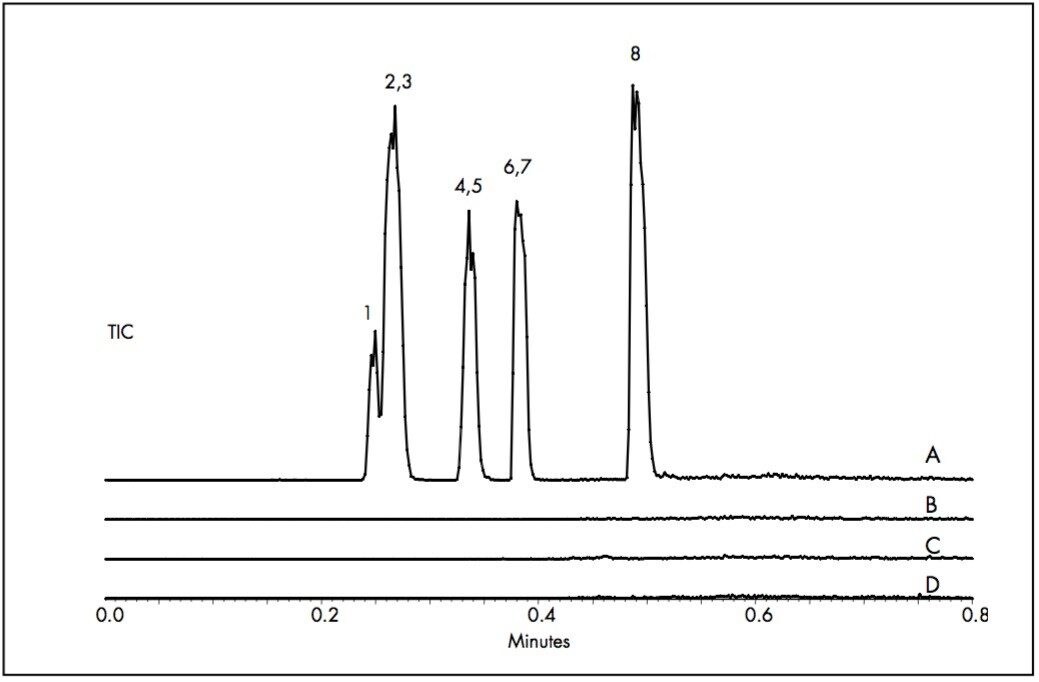
UltraPerformance Liquid Chromatography (UPLC Technology) in conjunction with ultra-violet (UV) detection can provide a high degree of assurance that the API residue is below the safety acceptance limit in a relatively short period of time (less than 5 minutes analysis time). In cases where UV is not sensitive enough, mass spectrometry (MS) is a useful addition for detecting low levels of residual drug substances.
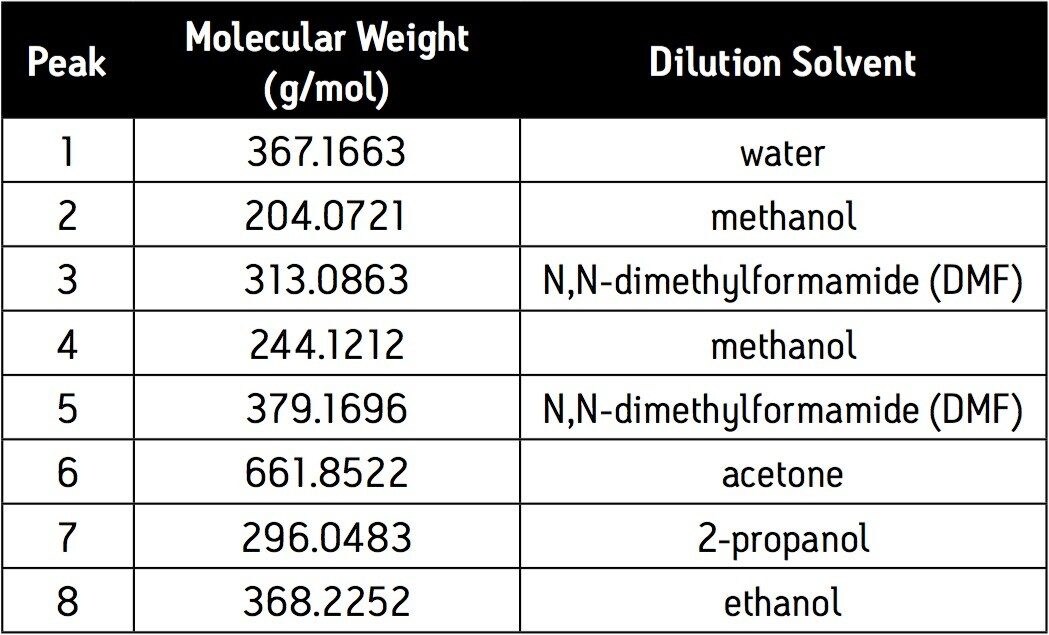
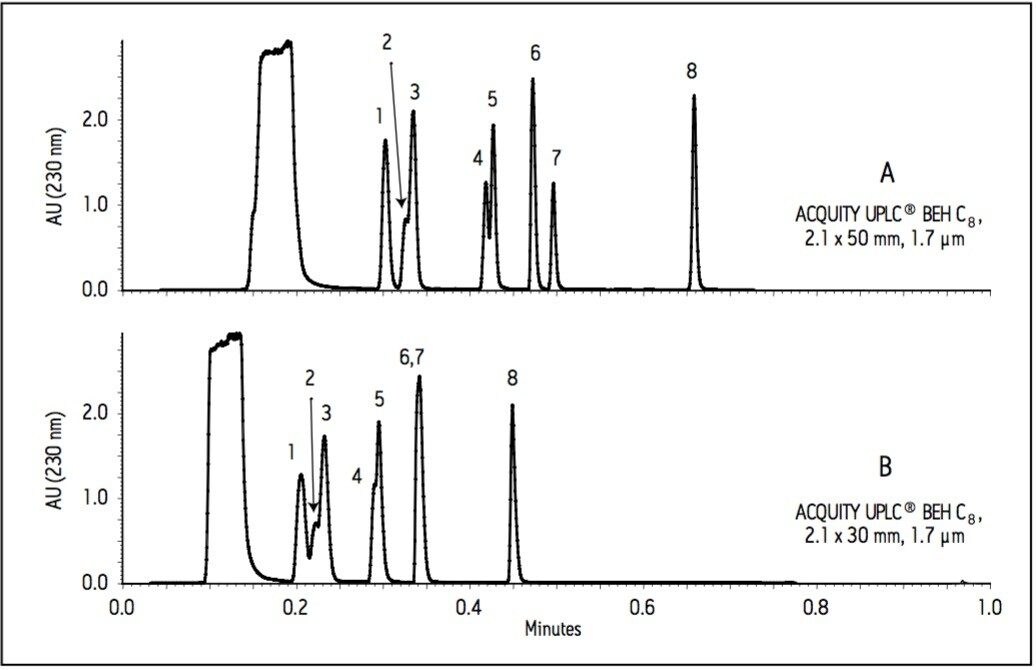
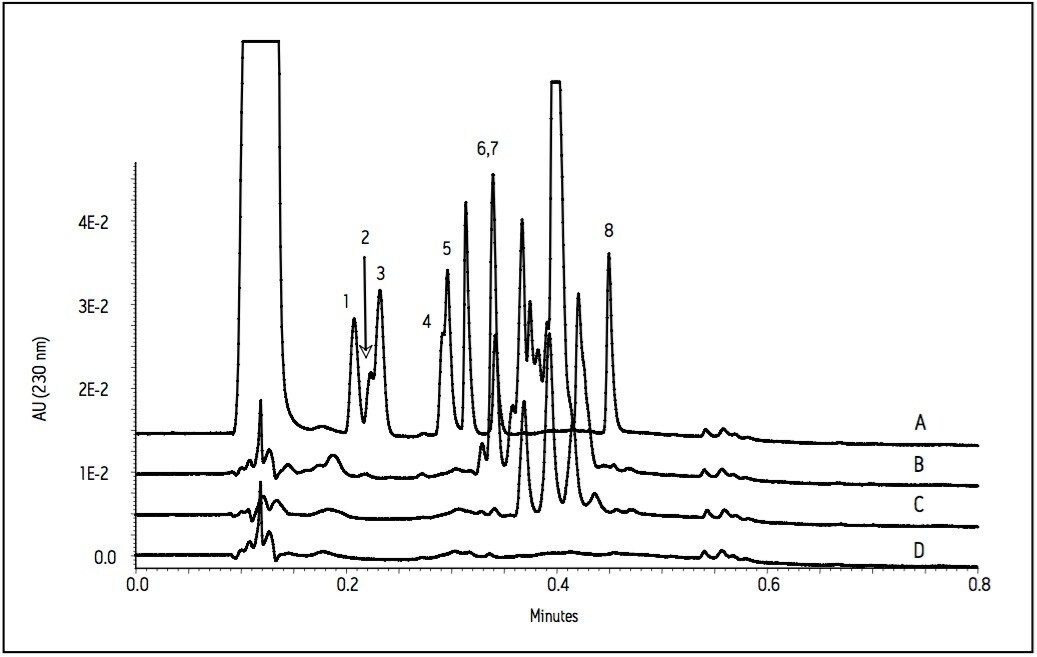
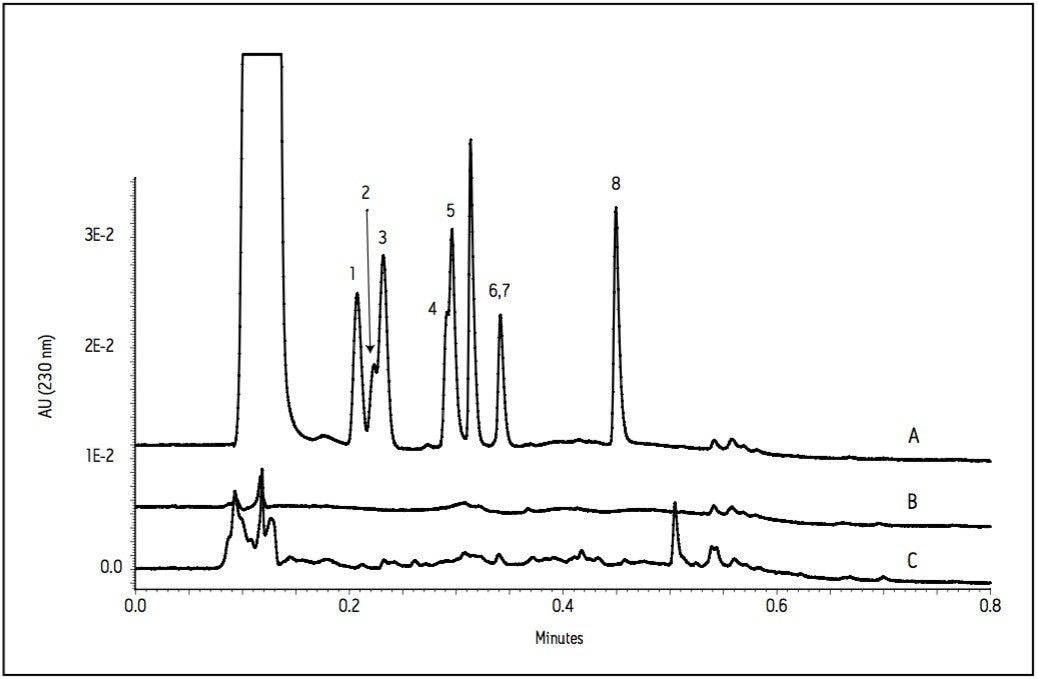
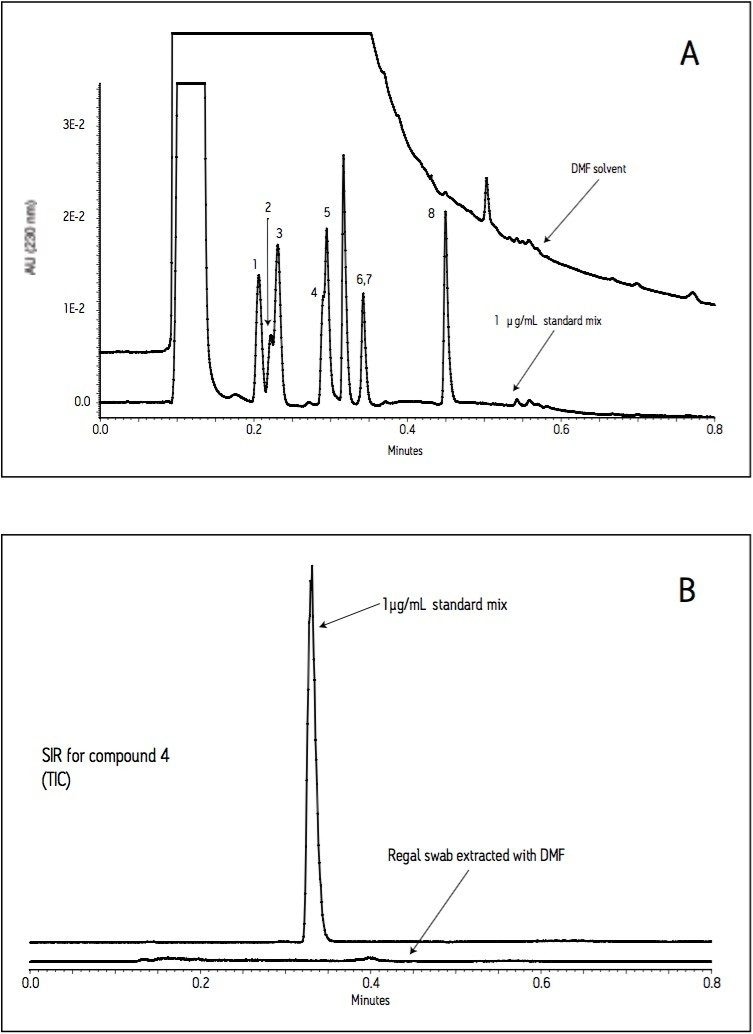
The current work highlights the use of UPLC-MS employing a short, 30 mm UPLC column along with a fast-scanning single quadrupole MS detector in order to support the cleaning validation procedures for eight APIs (Table 1). The customer desired a single UPLC analysis for all 8 compounds with a cycle time of less than 2 minutes. Previously, the customer had been using 8 different HPLC methods to measure drug residue levels after reactor vessel cleaning. This represents a significant reduction in quality control (QC) laboratory operating costs (i.e., less mobile phase preparation, less instrument down time), and an overall increase in productivity.
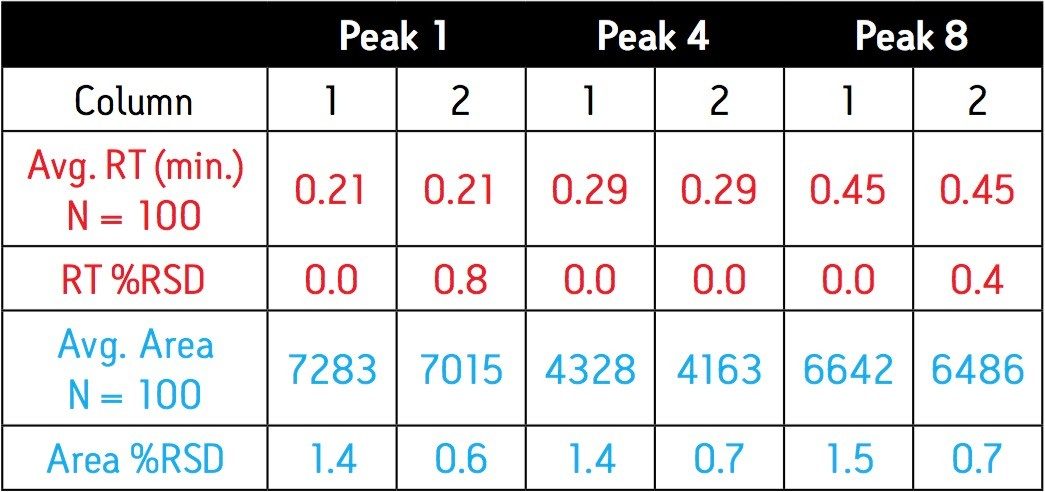
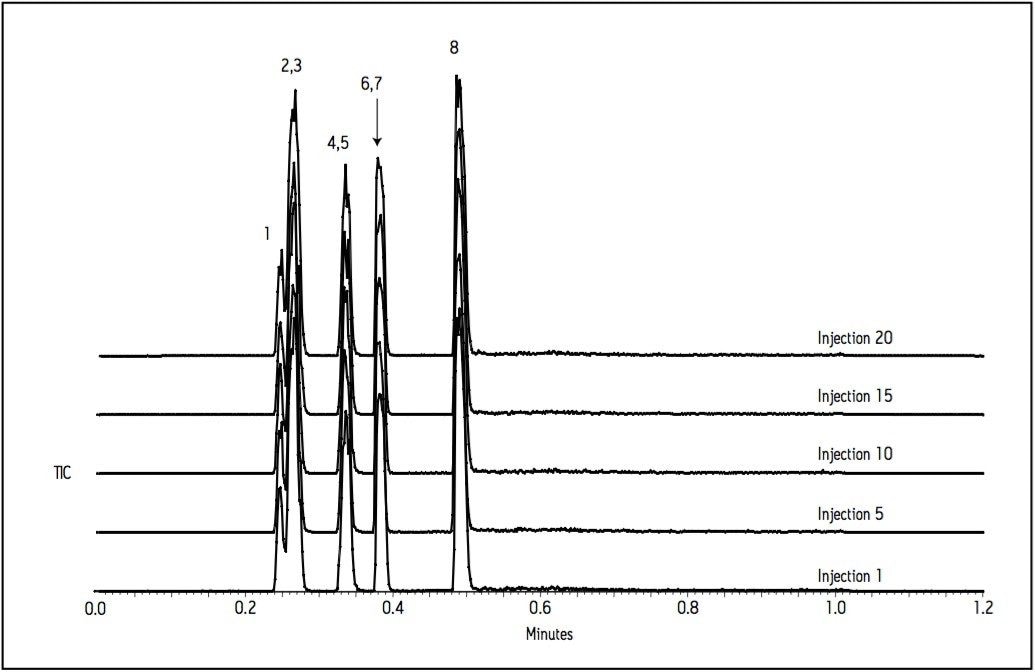
Since these APIs are synthesized individually, it is not necessary to resolve all compounds from each other in a single run. Repeatability in UV and MS must be determined, as well as the limit of detection (LOD) and limit of quantitation (LOQ). Finally, interferences from solvents and swabs used during cleaning of reactor vessels must be minimized in order to accurately measure API residue levels.