MALDI MS/MS is a powerful technique for the unambiguous identification and characterization of proteins. MS/MS is often used in conjunction with peptide mass fingerprinting to confirm the protein identification and characterize peptides not matched in the MS spectrum. When peptides are separated by only a few Daltons and their isotopic peak envelopes overlap, it is important in an MS/MS experiment to be able to set the precursor window of MS1, to transmit only the ions of interest and not isotopes from other peptides. Peptides with a similar m/z can often be observed with large proteins, or mixtures of proteins that, after enzymatic digestion, produce large numbers of peptides—some of which are likely to be similar in mass. If the precursor ion transmission window is too large, more than one ion will be transmitted and the MS/MS spectra will contain fragment ions from these other peptide species. This makes interpretation extremely difficult.
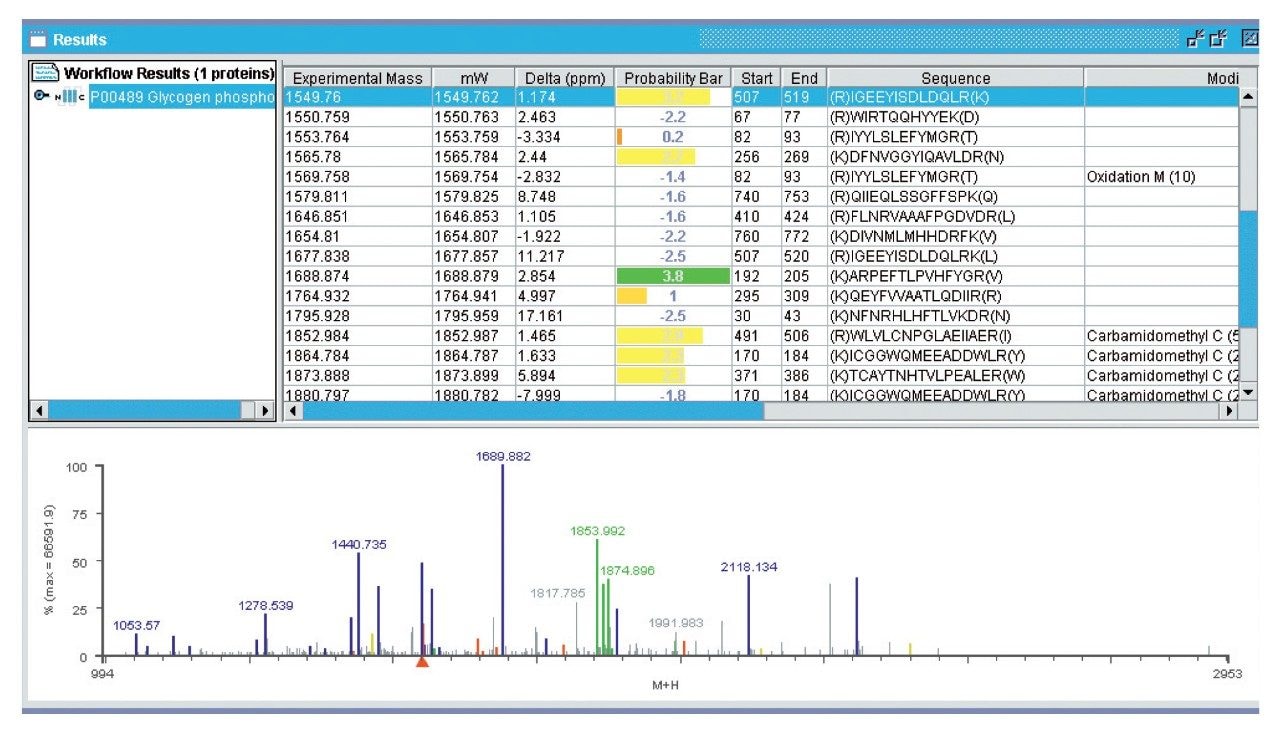
Peptides that do not match to protein sequences identified by peptide mass fingerprinting, are often chemically modified, contain amino acid substitutions or were produced by non-specific cleavage during the digestion process. Alternatively, unmatched peptides may occur when a mixture of proteins is present, some of which are at low levels or produce only a few tryptic peptides. Peptide mass fingerprinting normally relies on at least five matching peptides to confidently identify a protein.
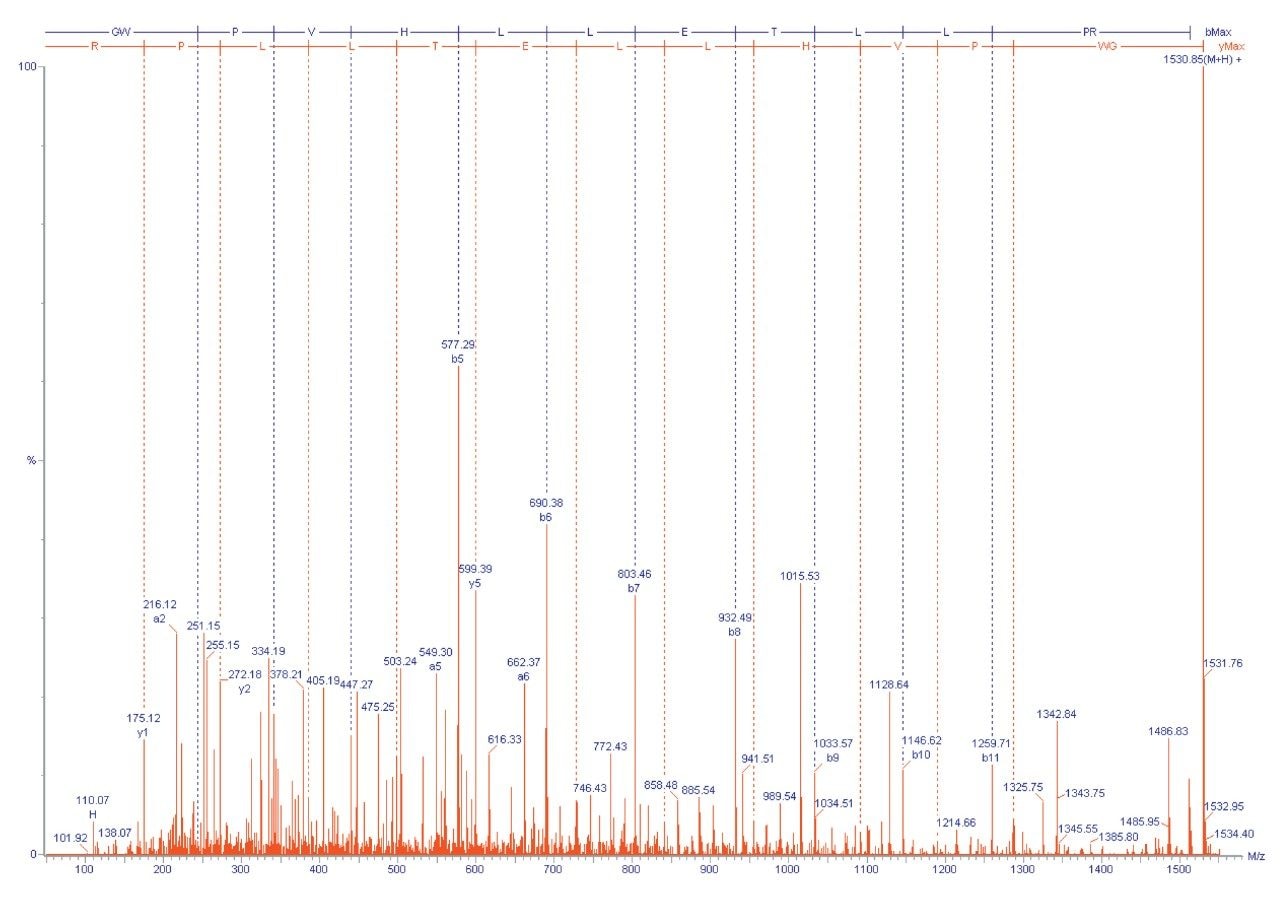
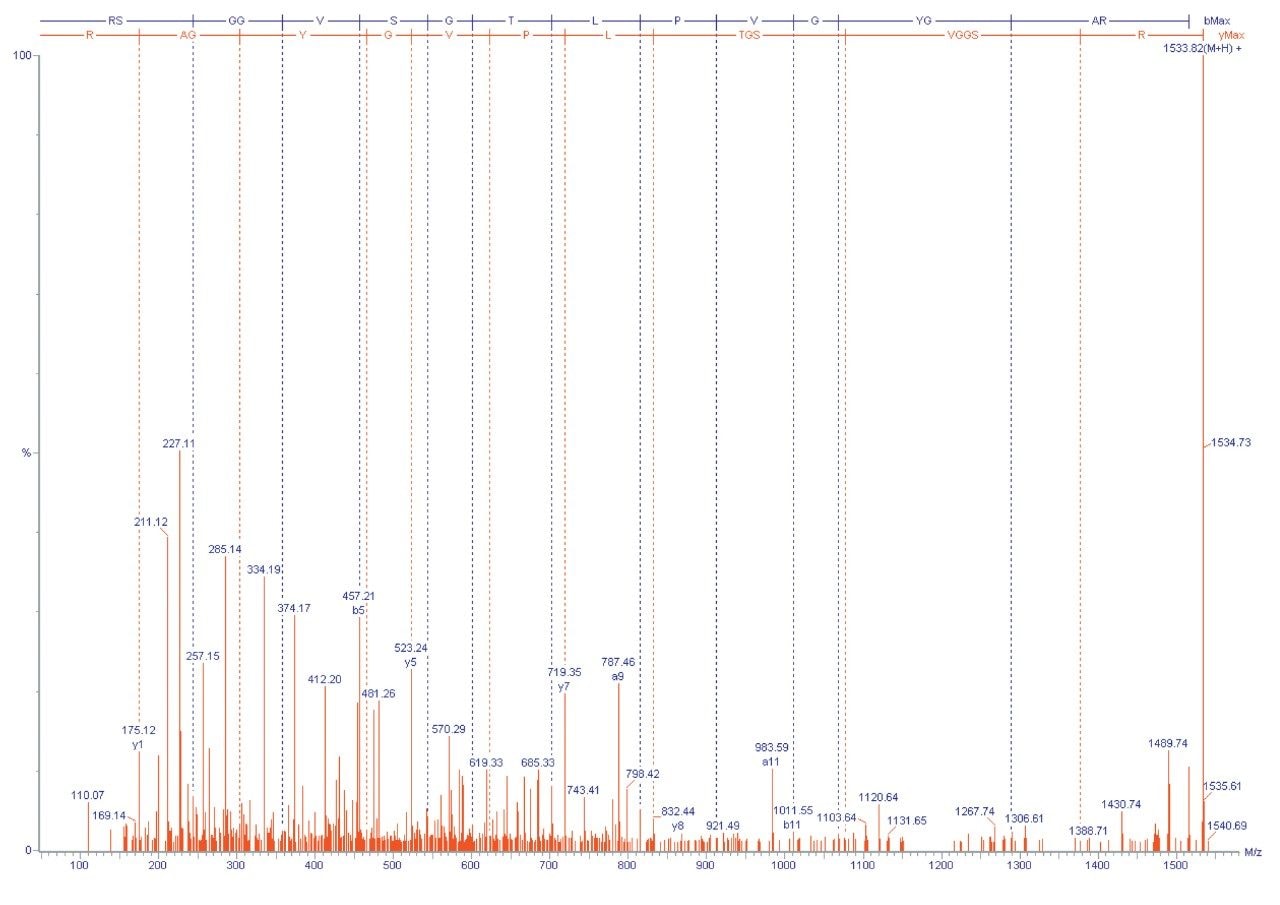
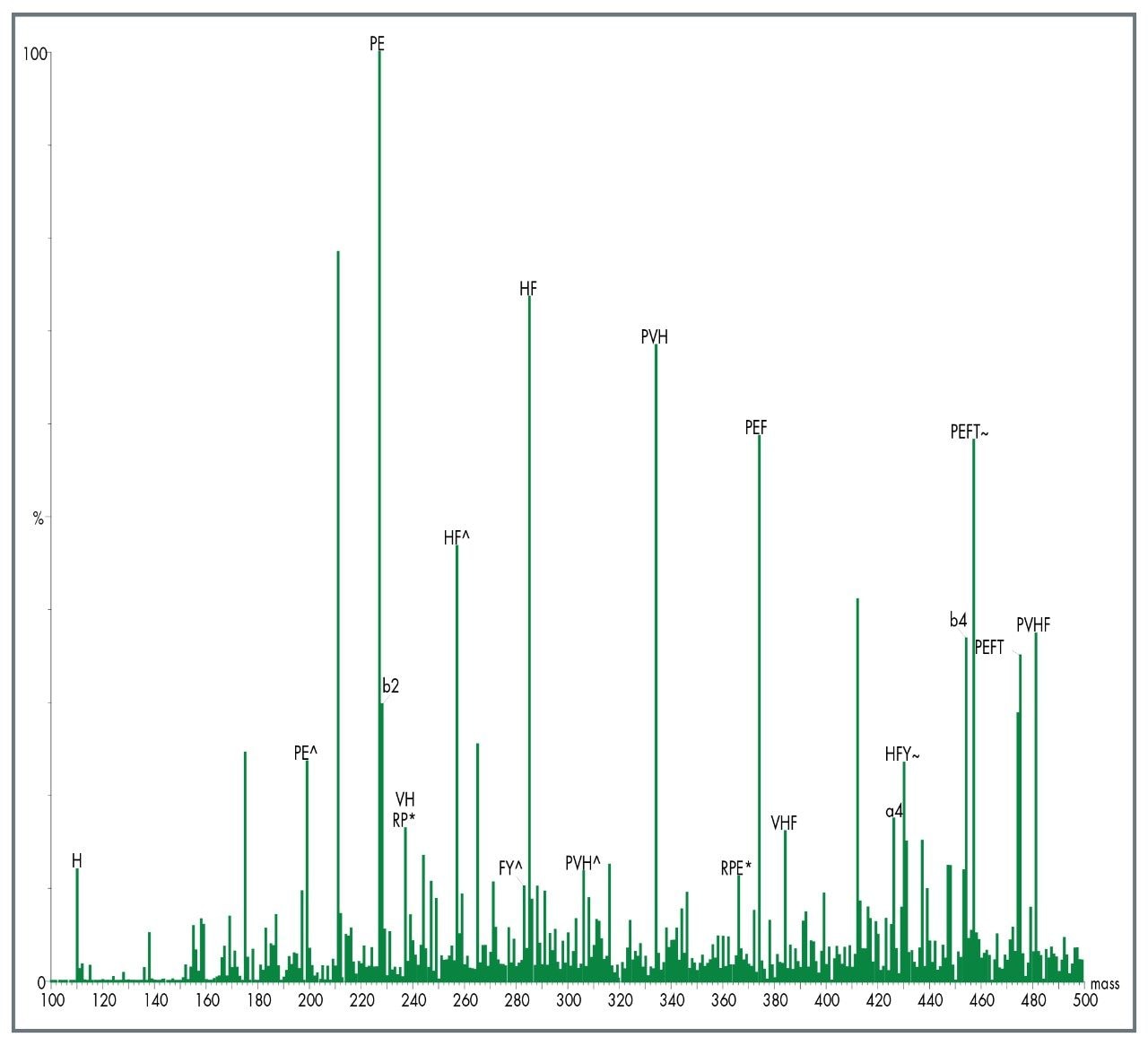
A different approach that can be applied is to fragment the unidentified peptide and compare the MS/MS fragment ion data with a sequence databank. One advantage of this approach is that proteins can often be identified from a single peptide. This is possible due to the specificity of exact mass MS/MS sequence information.
If a protein hit is still not identified then de novo sequence information can be obtained and compared with protein sequences in a databank using homology-based BLAST searching.
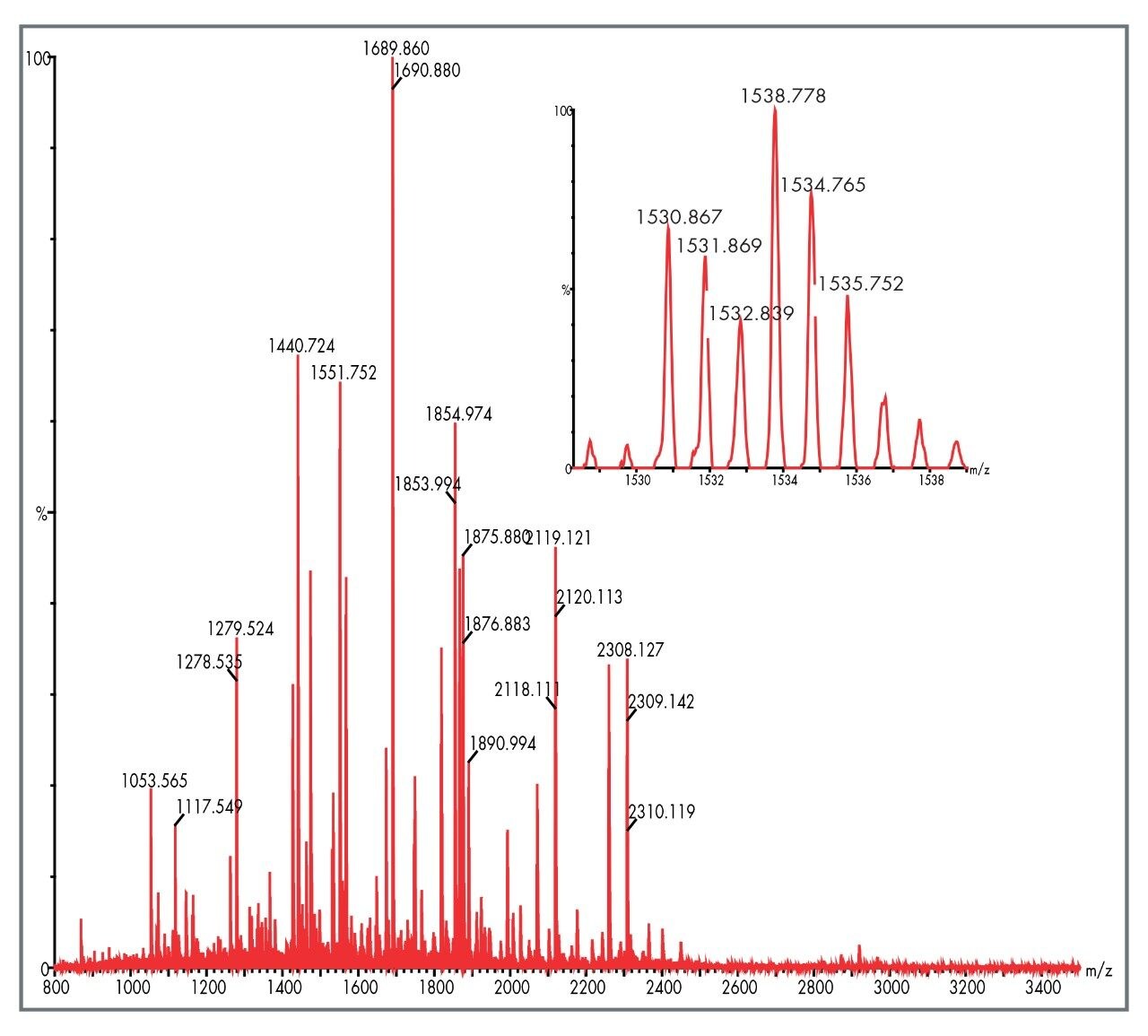
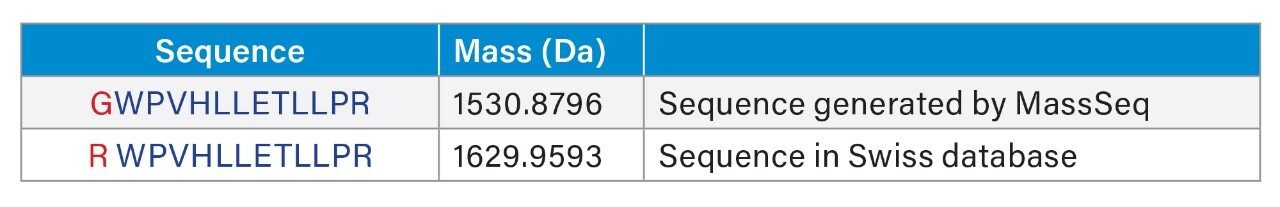
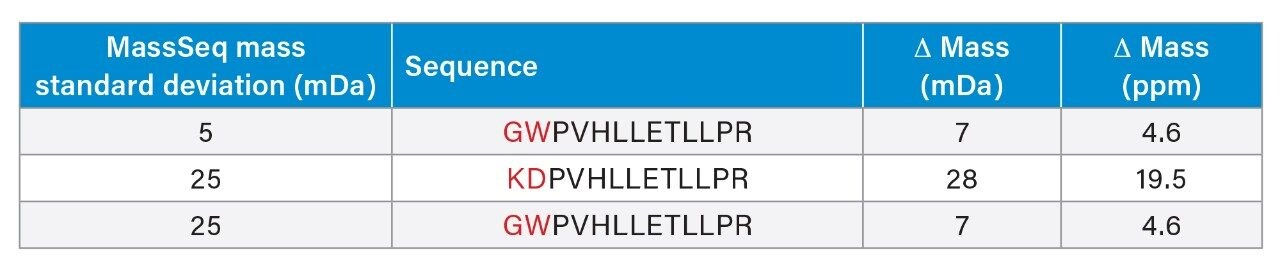
In this application note we highlight the need for accurate precursor ion selection windows, using as an example, two peptides from a tryptic digest of glycogen phosphorylase B that are not matched by peptide mass fingerprinting. These peptides at mass 1530 Da are only separated by 3 Da. We also discuss the advantages of having exact mass measurement for de novo sequencing of tryptic peptides from singly charged MALDI MS/MS data.