In metabolism studies, it is vital to understand how a particular drug is absorbed, distributed, metabolised, and eliminated by the body. Metabolite identification is a very important part of the drug discovery/development process, because early detection and identification of major and toxic metabolic routes can help to fine-tune drugs that go on to development. Metabolism is not an easy task and it can be extremely complex; that is why speed and accuracy of results are vital in order to make the right decisions at this stage.
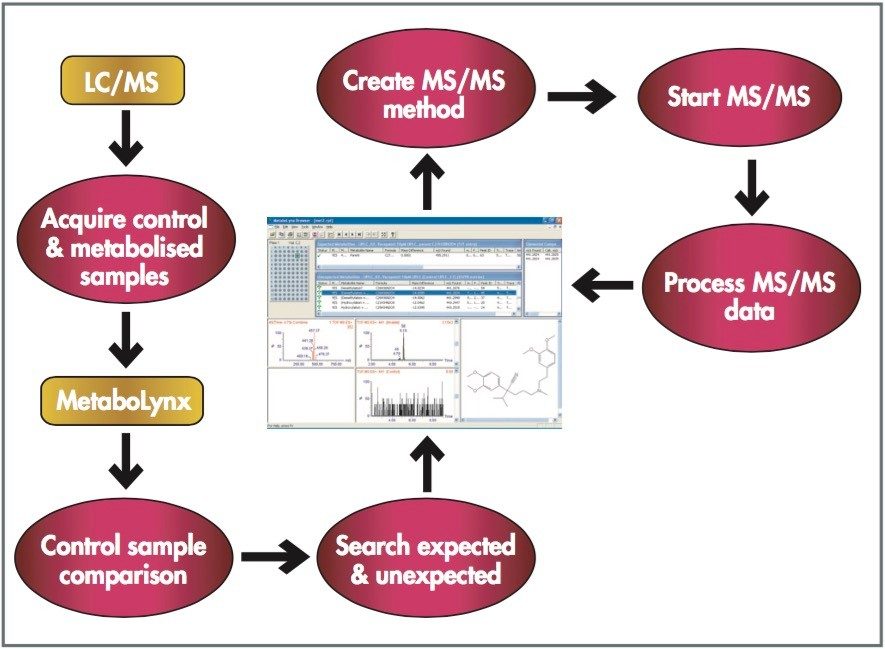
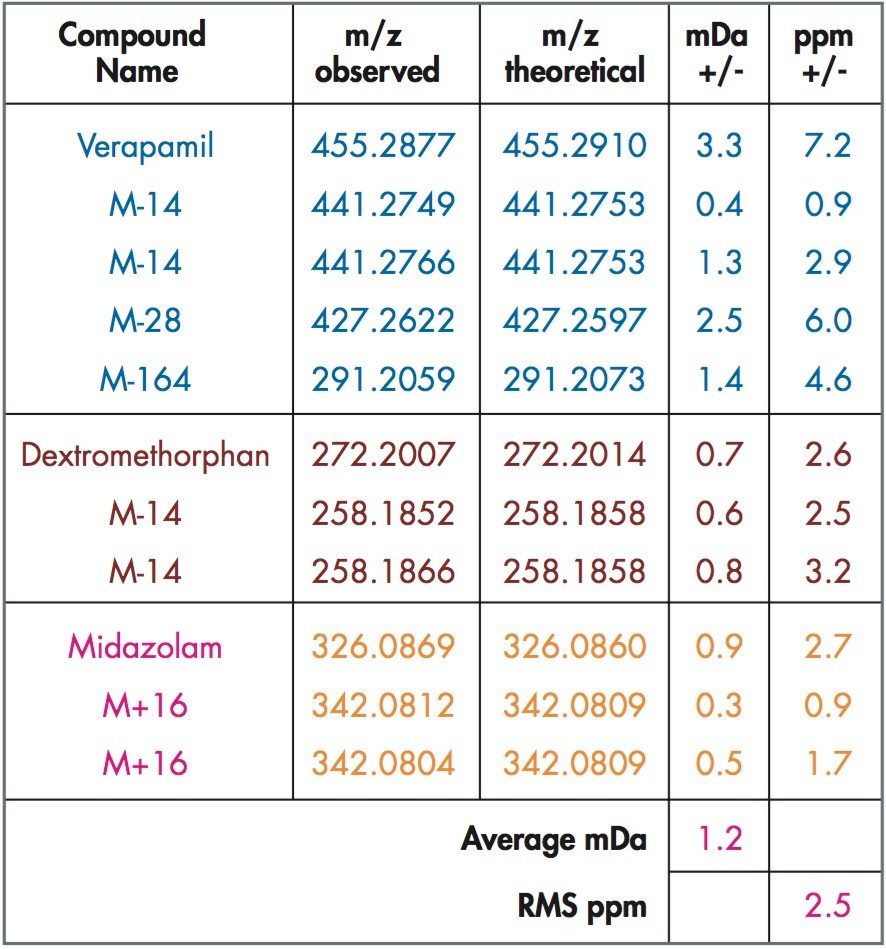
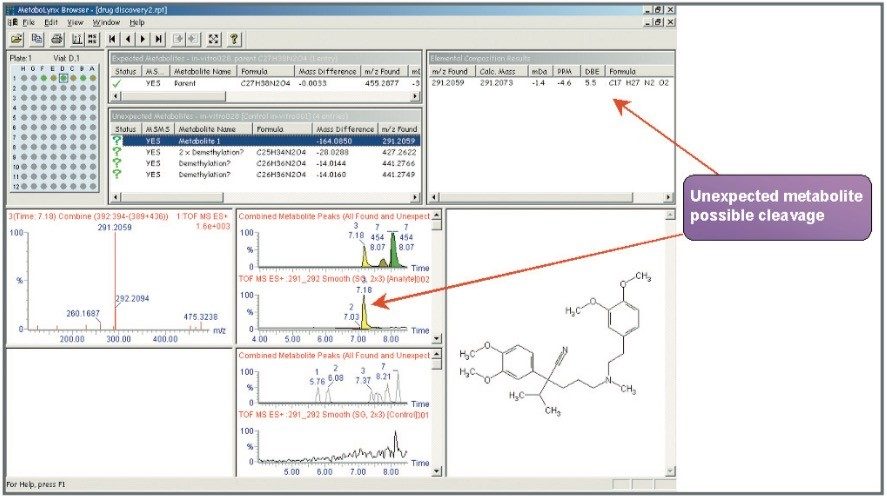
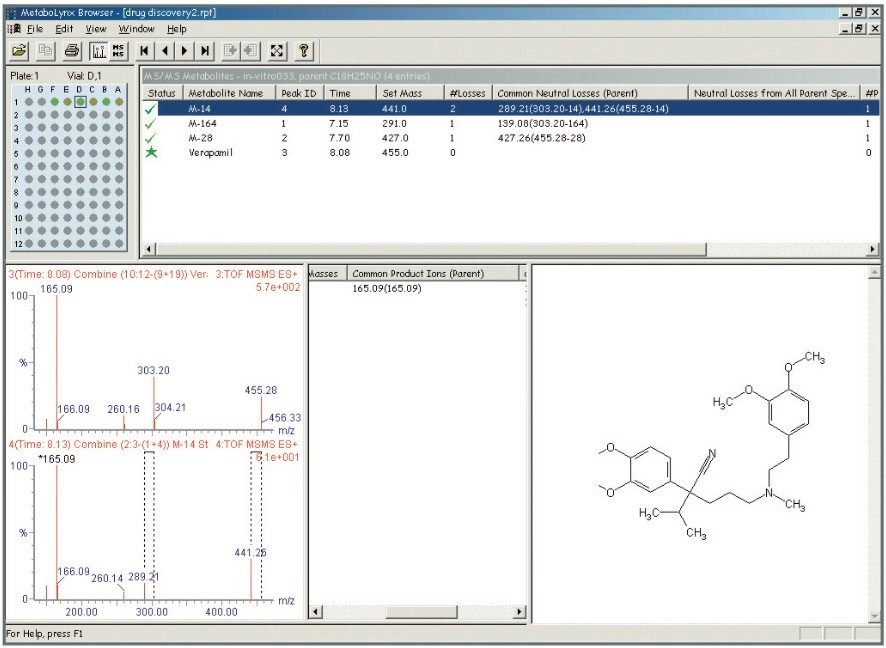
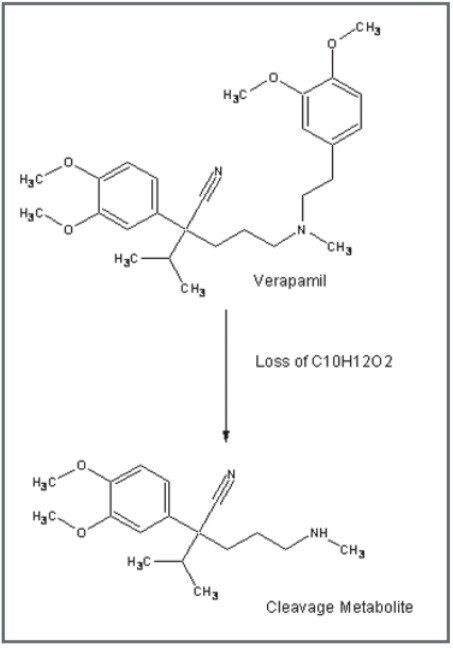
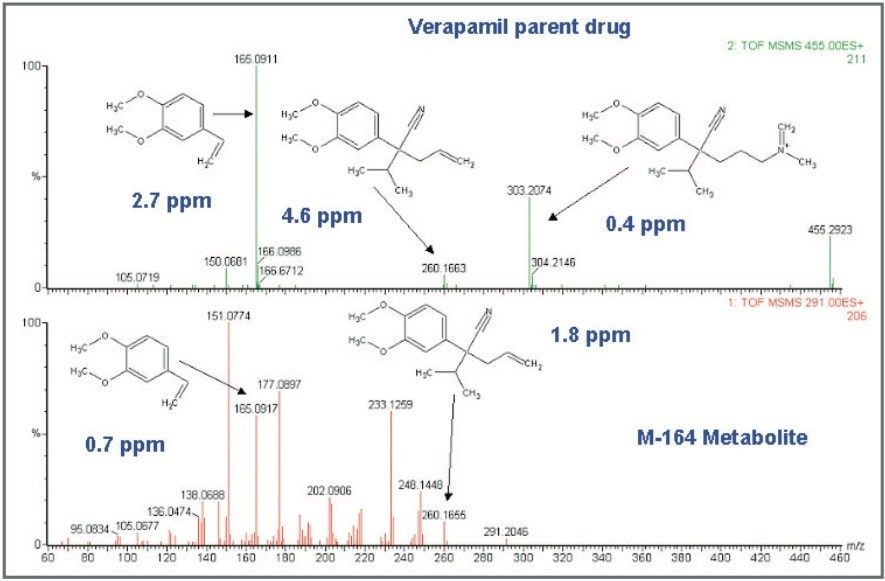
Mass spectrometry is a well established technique for pharmacokinetic and metabolism studies, due to the fact that it is sensitive, fast, and robust. Exact mass measurement technologies, such as quadrupole time of flight (Q-Tof) MS, offer accurate data and high sensitivity, which are very important for metabolite identification. Use of exact mass data in the determination of metabolite structures allows medicinal chemists to make the necessary “structural tuning” to achieve the desired compound biological activity. Moreover, as the demand to screen a large number of compounds in drug discovery increases, bottlenecks in data processing can often result. Therefore, there is a need to develop an automated approach that can cope with the number of candidates analysed in the drug discovery process. In this paper, we present an automated bioanalytical approach using exact mass in combination with an automated software package, MetaboLynx, for in vitro metabolite screening (Figure 1). A range of different compound incubates in microsomes at 5 μM will be shown. We will show that the MetaboLynx Software algorithm enables the identification of expected and unexpected metabolites from a control sample comparison. Moreover, we will also show how we can automate the entire process of metabolite detection and MS/MS method creation for subsequent sample analysis for identification purposes. Finally, we will show how enhanced mass measurement accuracy in the assay results may increase sample throughput and improve decision-making steps. This is especially applicable in the discovery phase, where some “compound structural tuning” may be required to achieve the pharmacokinetic and metabolism effects required for a specific novel structure.