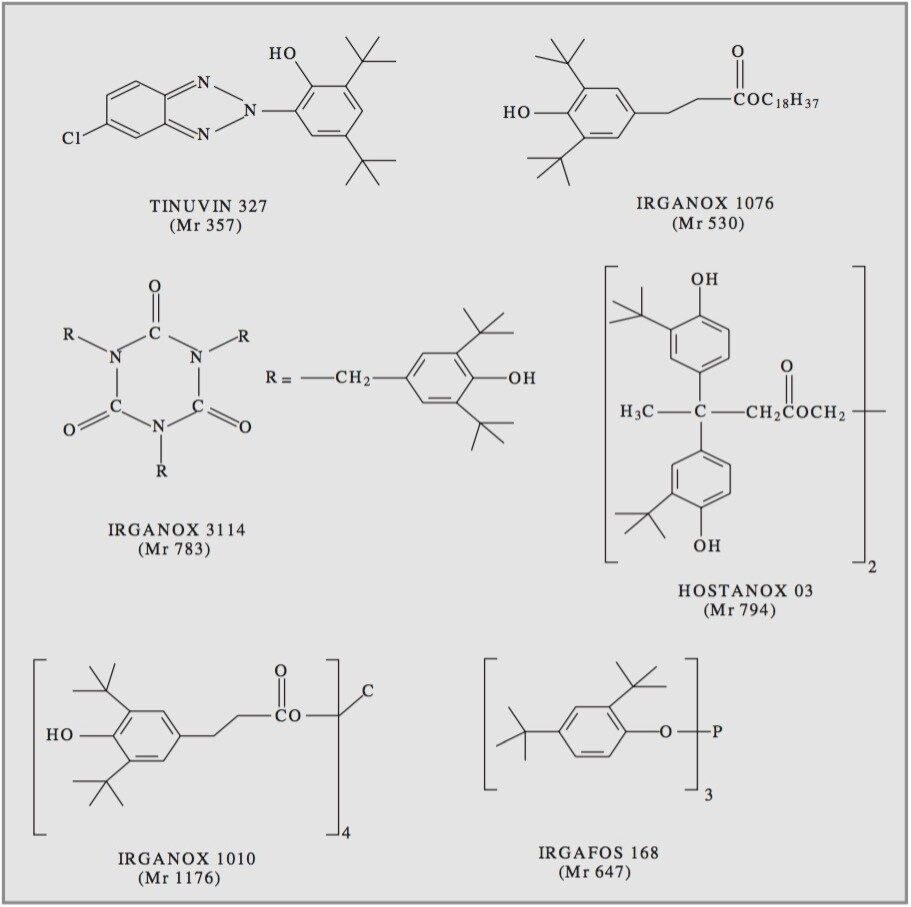
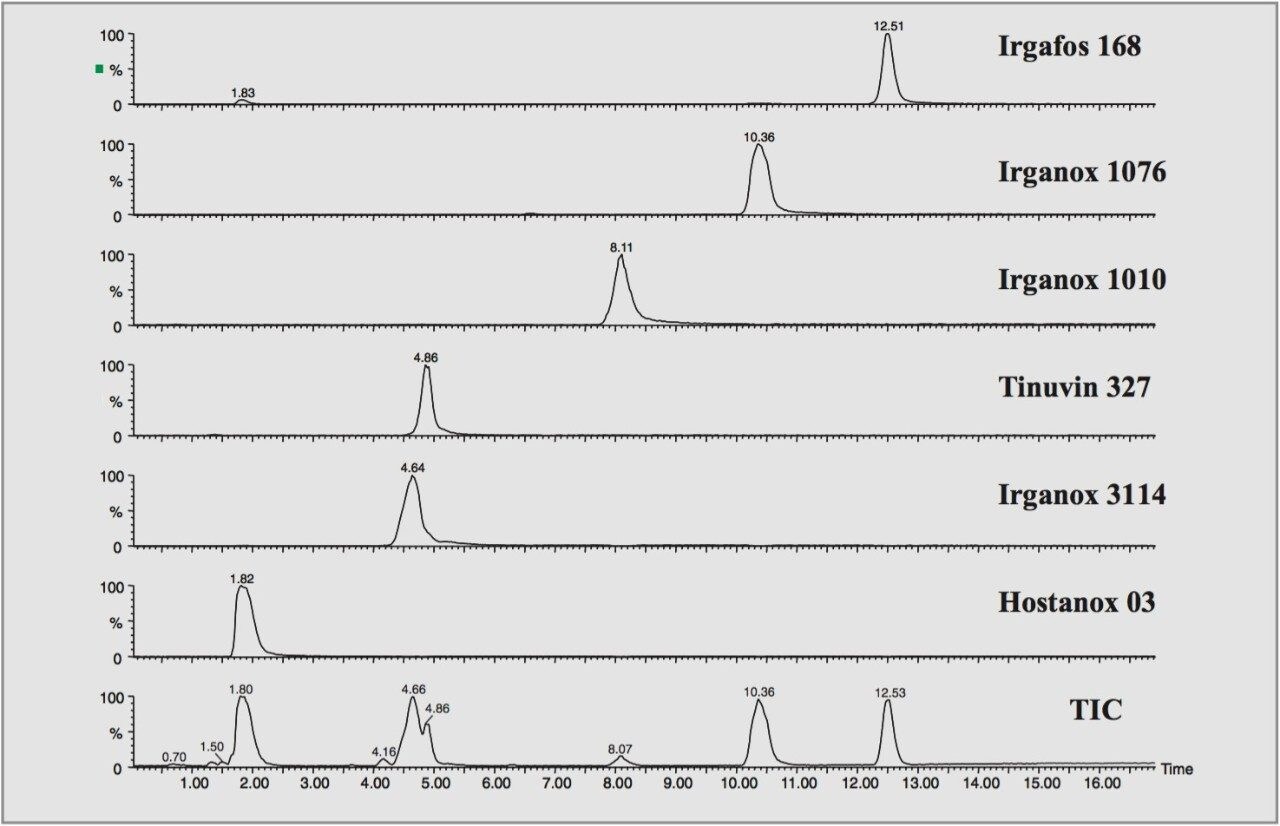
Polymer additives have several key functions. They aid processing and finally characterize the formulated commercial product within the field of polymer chemistry. Typical additives act, for example, as light stabilizers, antioxidants, flame retardants or plasticizers.
Polymer additive analysis is important because of possible health and environmental risks that can arise from the manufacture of certain plastic materials, which are intended to come into contact, for example, with foodstuffs.1
The possibility of migration of the additives into foodstuffs needs to be carefully monitored and therefore the levels of additives in the plastic need to be determined and well regulated.
Often added at levels between 0.1–3% w/w, the determination of the fate of these additives during and after use, their presence in competitor products and inherent purity presents a daunting analytical challenge. Therefore, precise identification is a crucial requirement in their characterization. Synthetic polymer formulations are complex, therefore extraction procedures from the polymer matrix usually precede chromatographic separation.
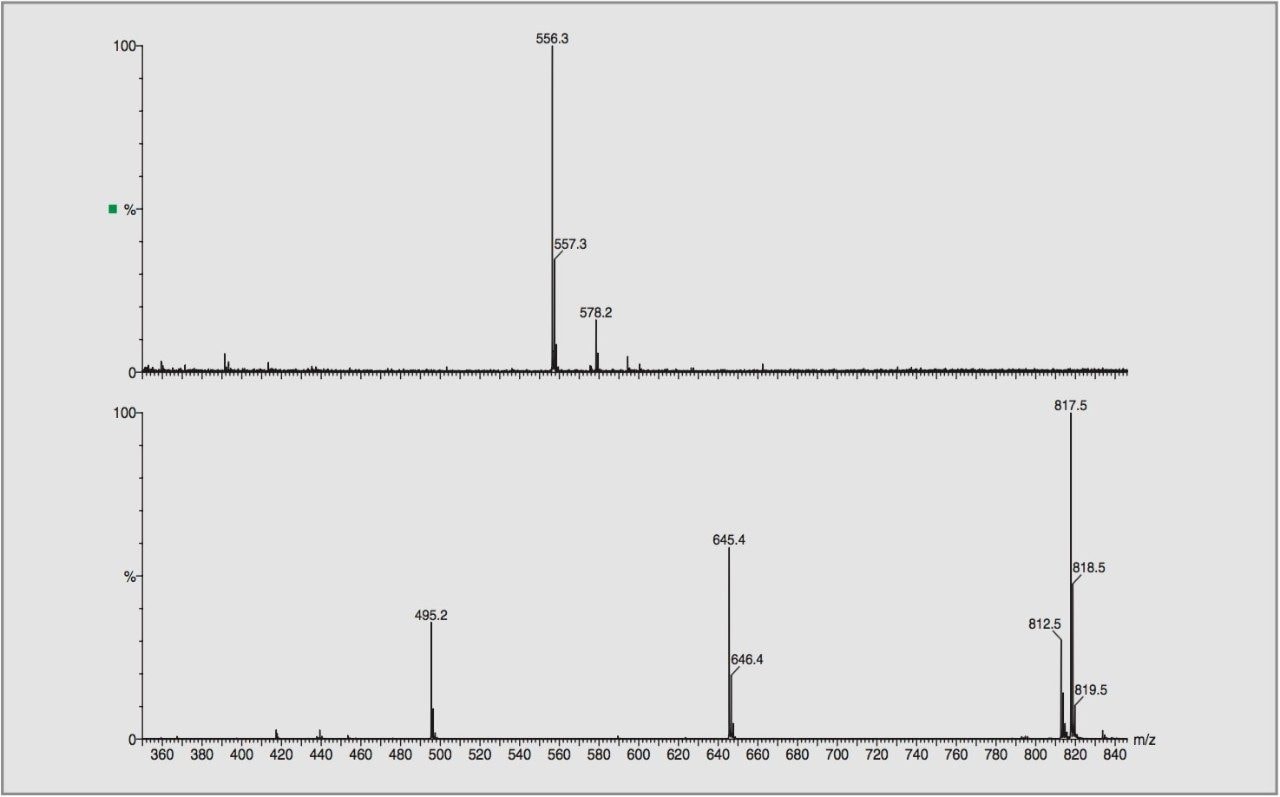
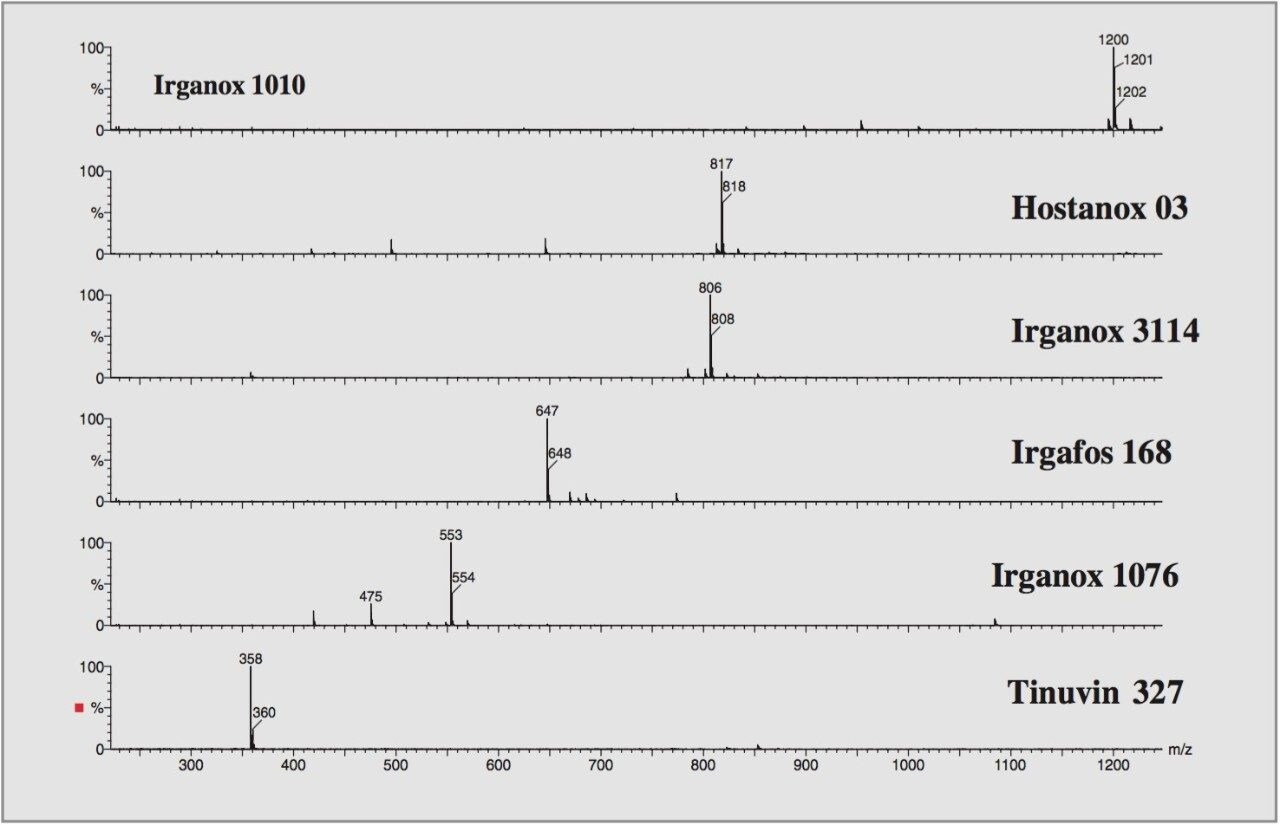
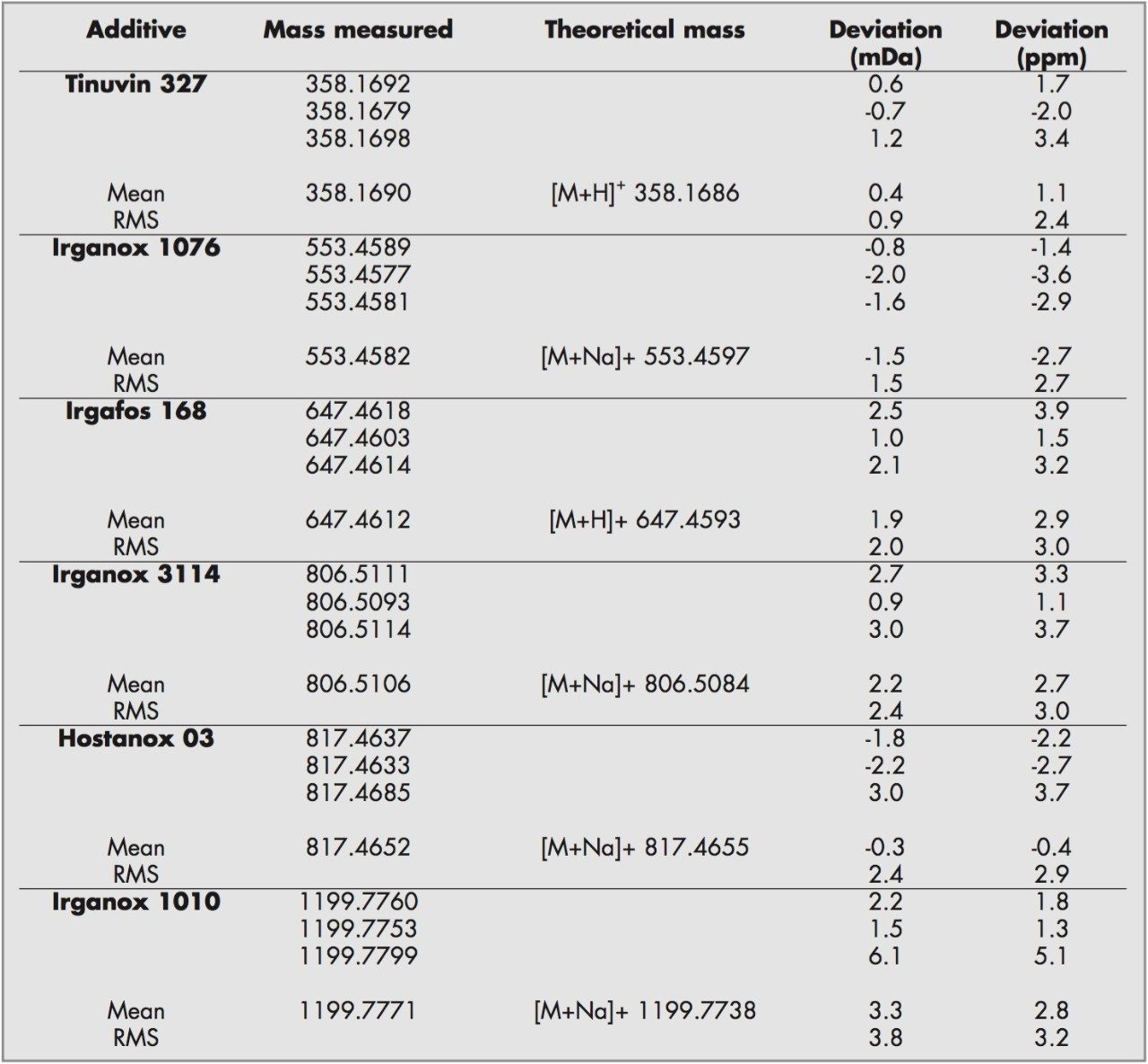
Electrospray mass spectrometry and tandem mass spectrometry have been previously employed on some of the additives studied.2 Here high and low collision energies were used to produce nominal mass product ion spectra. From the results mechanistic and fragmentation pathways were proposed.
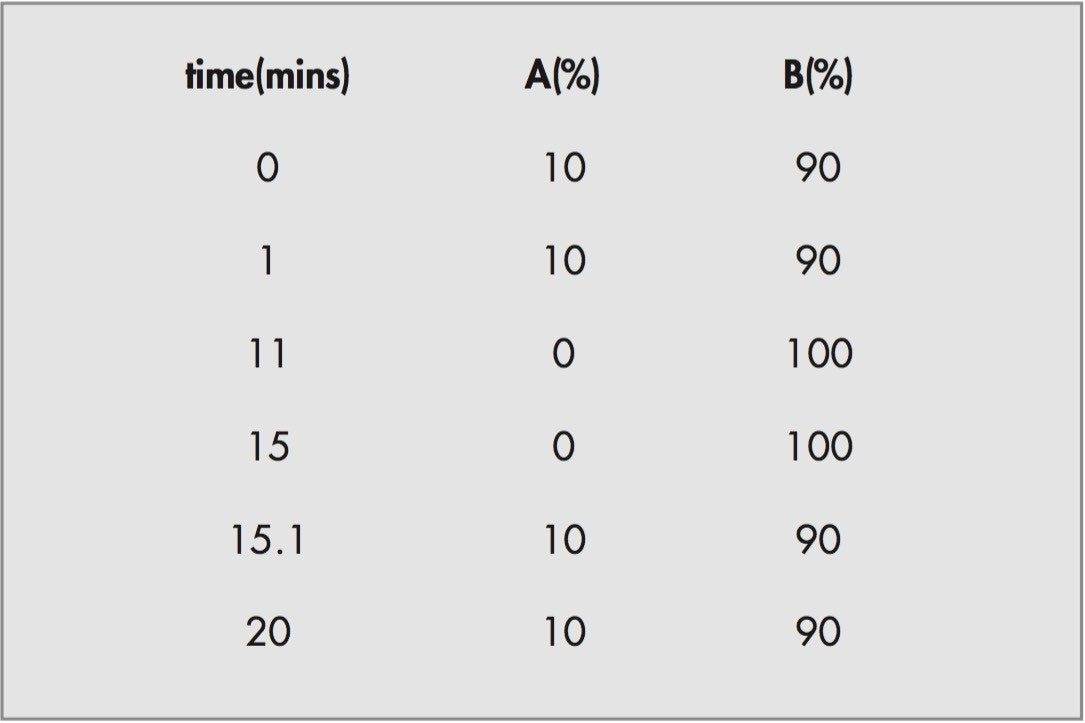
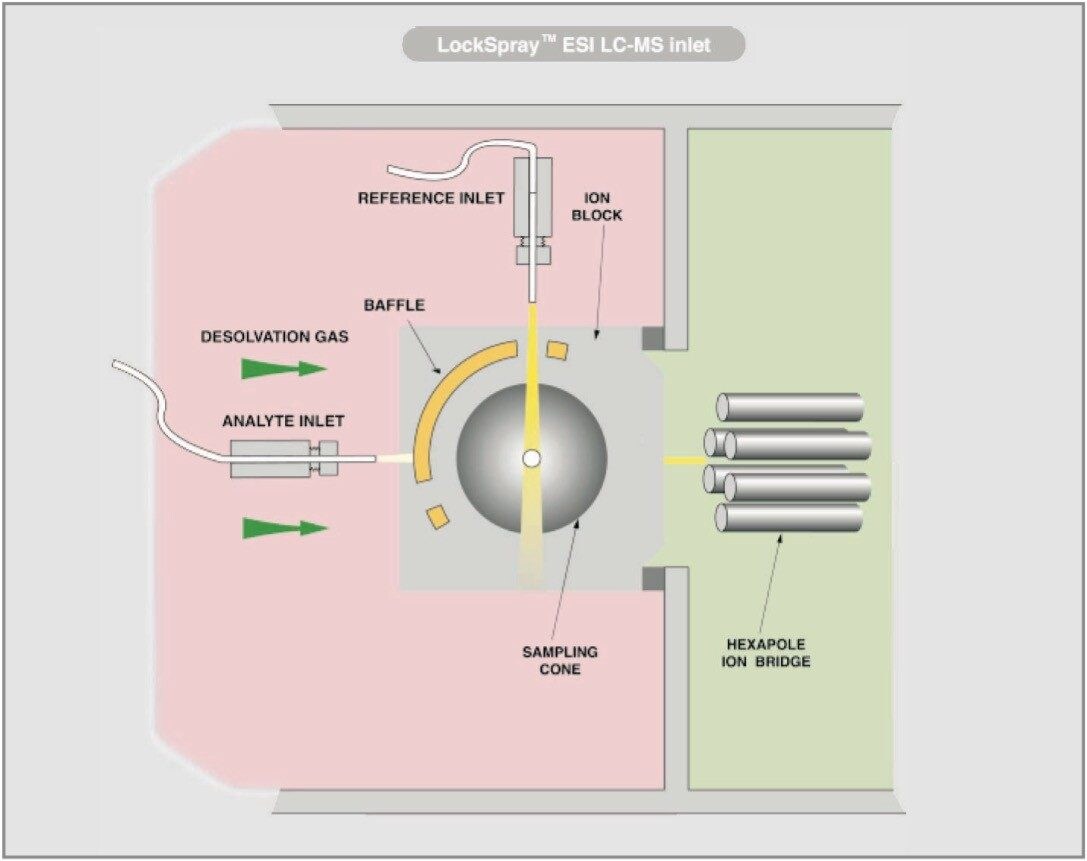
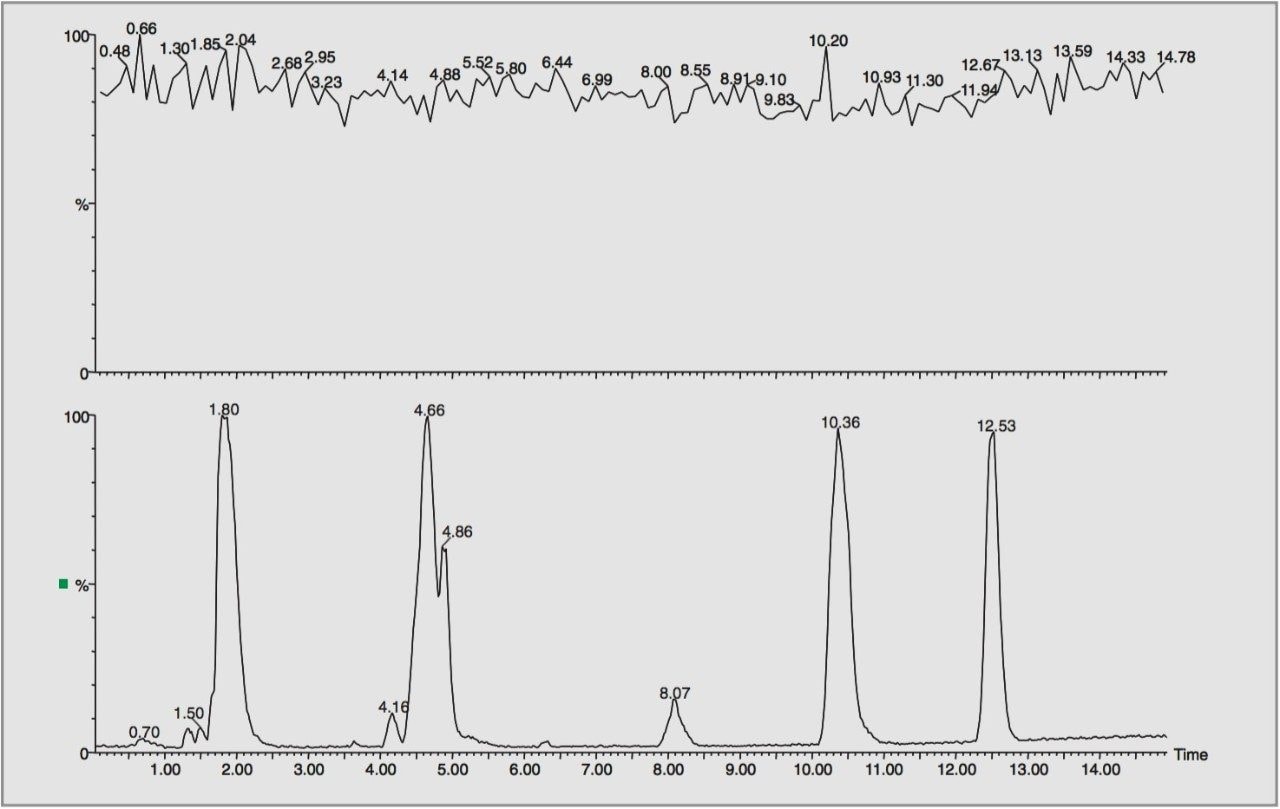
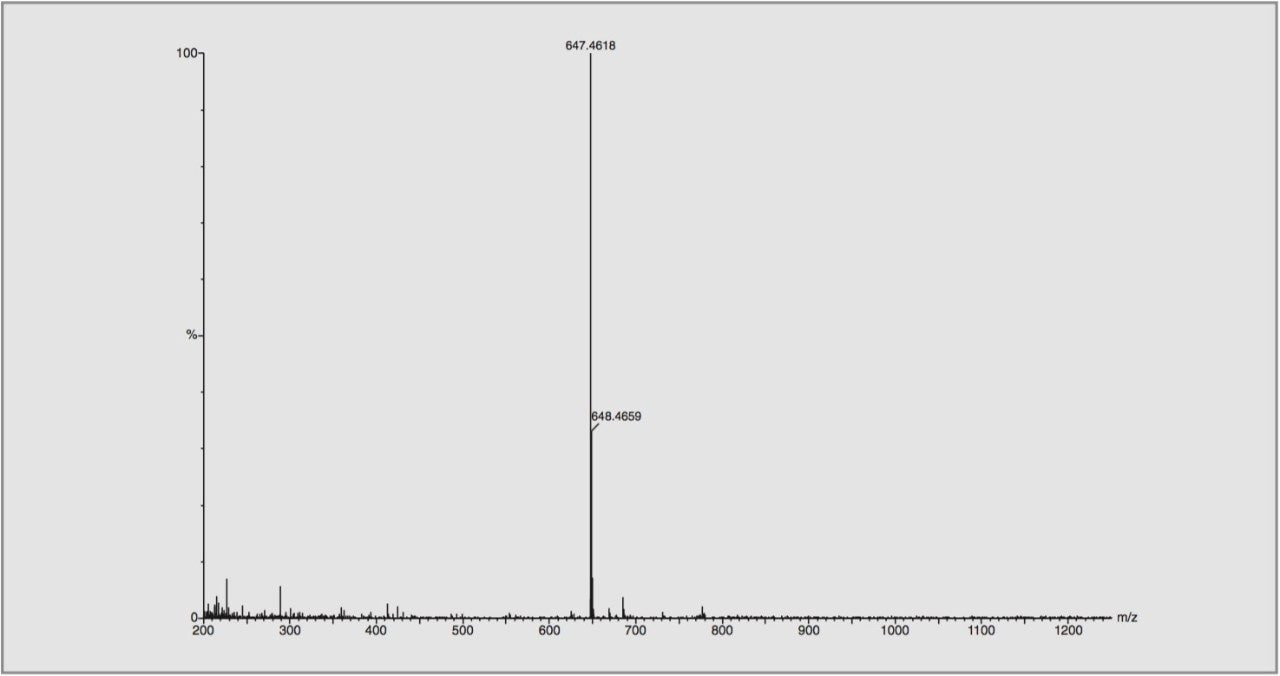
For this study, oa-Tof-MS has been conveniently used with liquid chromatography for the exact mass measurement of the same group of additives previously investigated.2