ACQUITY™ QDa™ II 質量検出器を用いたアトルバスタチン中の不純物分析の合理化による、検出および定量の向上
要約
- アトルバスタチン API の不純物のルーチン分析で、UV クロマトグラフィーワークフローを補完するための QDa II 質量検出器の実際のアプリケーションを実証
- ICH-Q3 ガイドラインによる適格性評価しきい値 0.15% 以下の一部の不純物を測定する分析ワークフローの適合性を評価
- 未知化合物の構造特性に関する洞察を深めるための、イオン源内フラグメンテーションのメリットを明確に提示
- 規制対象の製薬環境での規制準拠した不純物試験における Empower™ クロマトグラフィーデータシステム(CDS)の使いやすい機能およびメリットを紹介
はじめに
アトルバスタチンは、高コレステロール血症などの症状の管理や、心血管疾患のリスク低減のために、世界中で最も幅広く処方されている医薬品の 1 つです。アトルバスタチンには LDL コレステロール値を低減する効能があるため、世界中の何百万人もの患者に提供されている優れた医薬品です。一方、医薬品製剤中の不純物の存在が、患者の安全性に重大な懸念を及ぼす可能性があります。このような不純物は、活性物質の合成プロセスに起因して、あるいは特定の環境条件下での分解生成物として生じることがあります。また、最終的な医薬品の安全性および有効性を損なう可能性がある毒物学的特性や薬理学的特性を有している場合があります。
アトルバスタチンは通常、活性ヒドロキシル酸のカルシウム塩として、10 ~ 80 mg/日の用量で投与されます1。 ICH-Q3 ガイドラインによると、1 日の最大用量が 2.0 g 未満の医薬品は、不純物に関する適格性評価しきい値を 0.15% にする必要があります2。
活性物質としてのアトルバスタチンに関する欧州薬局方のモノグラフによると、不純物について、関連不純物 A および B の場合 0.3%、不純物 C および D の場合 0.15%、その他の特定されていない不純物の場合 0.1% という限度が指定されています3。
不純物試験の従来の方法、つまり UV 検出と組み合わせたクロマトグラフィー手法は、医薬品分析における品質管理(QC)の基本になっています。このワークフローに質量検出を補うことで、不純物の確実かつ迅速な同定、特性解析、定量が可能になります。さらに、質量検出では、非常に低い検出限界周辺の濃度であっても、不純物の同定に役立つ分子量情報も得られることで、分析の範囲を広げることができます。
このアプリケーションでは、アトルバスタチン API の関連不純物の迅速で正確な同定および定量における質量検出の適合性を実証することを目的としています。さらに、疑似 MS/MS アプローチを使用して、イオン源内フラグメンテーションを通じて不純物を特性解析することにより、不純物分析用の包括的ソリューションを紹介することを目的としています。
実験方法
アトルバスタチンカルシウム塩および 3 種類の関連不純物(不純物 A、C、I)の市販のサンプルは、Sigma Aldrich から購入し、分析を行うまで 4 ℃ で保管しました。サンプルをメタノール(MeOH)に溶解し、分析用に 40:60 H2O:MeCN の溶媒混合液で段階的に希釈しました。
LC-MS の実験条件
|
LC システム: |
ACQUITY UPLC™ H-Class システム(FTN-R サンプルマネージャ搭載) |
|
検出: |
ACQUITY QDa II 質量検出器 |
|
UV システム: |
フォトダイオードアレイ(PDA)検出器 |
|
カラム: |
ACQUITY UPLC CSH™ Phenyl-Hexyl カラム、1.7 µm、2.1 × 100 mm(製品番号:186005407) |
|
カラム温度: |
30 ℃ |
|
サンプル温度: |
10 ℃ |
|
注入量: |
2 µL |
|
流速: |
0.4 mL/分 |
|
実行時間: |
27.5 分 |
|
移動相 A: |
10 mM 酢酸アンモニウム水溶液 |
|
移動相 B: |
0.1% ギ酸アセトニトリル溶液 |
|
バイアル: |
透明ガラス 12 × 32 mm スクリューネックバイアル(製品番号:186000273) |
|
イオン化: |
ポジティブエレクトロスプレー(ES+) |
|
キャピラリー電圧: |
1.1 kV |
|
脱溶媒温度: |
600 ℃ |
|
イオン源温度: |
120 ℃ |
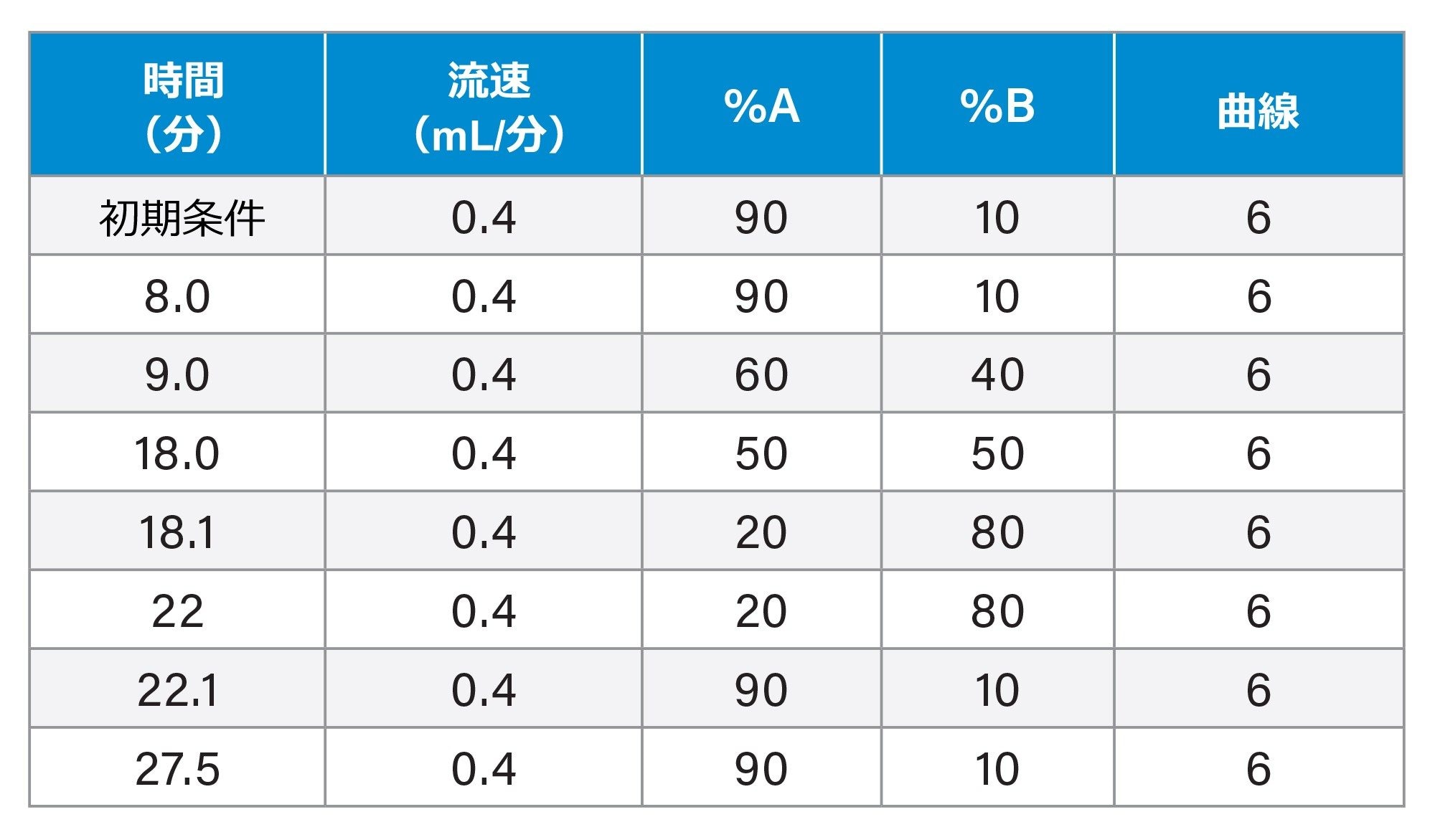
LC グラジエント
ソフトウェア
|
データ取り込み、解析、レポート作成: |
Empower CDS 3.0 |
結果および考察
分析法開発
このアプリケーションで紹介する分析法は、欧州薬局方のモノグラフによって規定されている分析法と異なり、移動相の一部として(毒性と不安定性の見られる)テトラヒドロフラン(THF)を使用せずに開発されたものです4。 代わりに、サンプル分析全体で pH を下げてピーク形状を改善するために、水系移動相に酢酸アンモニウムバッファーを添加し、有機移動相にギ酸を添加しています。移動相を pH 4.0 ~ 4.2 に最適化することを含め、複数の LC カラムと移動相組成も試験しましたが、クロマトグラフィーピークの分離度は、最終的な分析法と比較して改善していませんでした5。 最終的に、記載されている分析法は、欧州薬局方のモノグラフ分析法よりも約 60 分短く、注入あたりに使用する溶媒量は 10% 未満になっています。
この分析法を、アトルバスタチンと一部の不純物(A、C、I)の 1 µg/mL 混合液を使用して最適化しました。コーン電圧、キャピラリー電圧、脱溶媒温度がすべて最適化されました。文献に記載されている分析法のほとんどでは C18 カラムを選択していますが、CSH Phenyl-Hexyl カラムが、アトルバスタチン化合物と関連不純物 C の間の分離を改善するのに最適であることがわかりました。API のピークテーリングを低減するには、カラム温度 30 ℃ が最適でした。
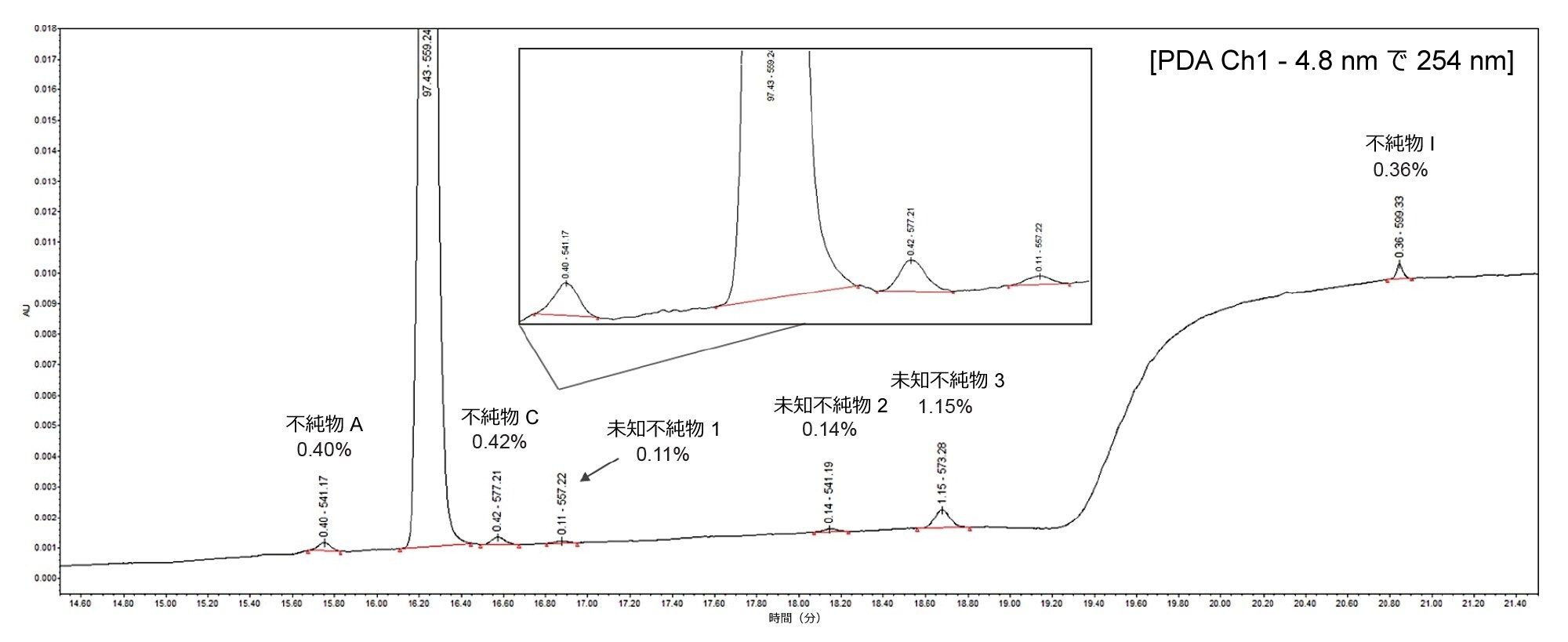
まず、分析法の性能を評価するために、25 µg/mL のアトルバスタチンサンプルに対して、この API に対する相対濃度範囲が 0.36 ~ 0.42% になるように不純物をスパイクしました。図 1 で未知不純物 1、2、3 とラベル付けした 3 種類の未知不純物は、PDA のトレースで、API に対する相対量がそれぞれ 0.11、0.14、1.15% と定量されました。
直線性
直線性を評価するため、API および不純物混合標準試料(不純物 A、C、I を含む)の両方について、連続希釈を行いました。すべての検量線は記載している分析法を使用して取得したもので、合計実行時間は 27.5 分でした。
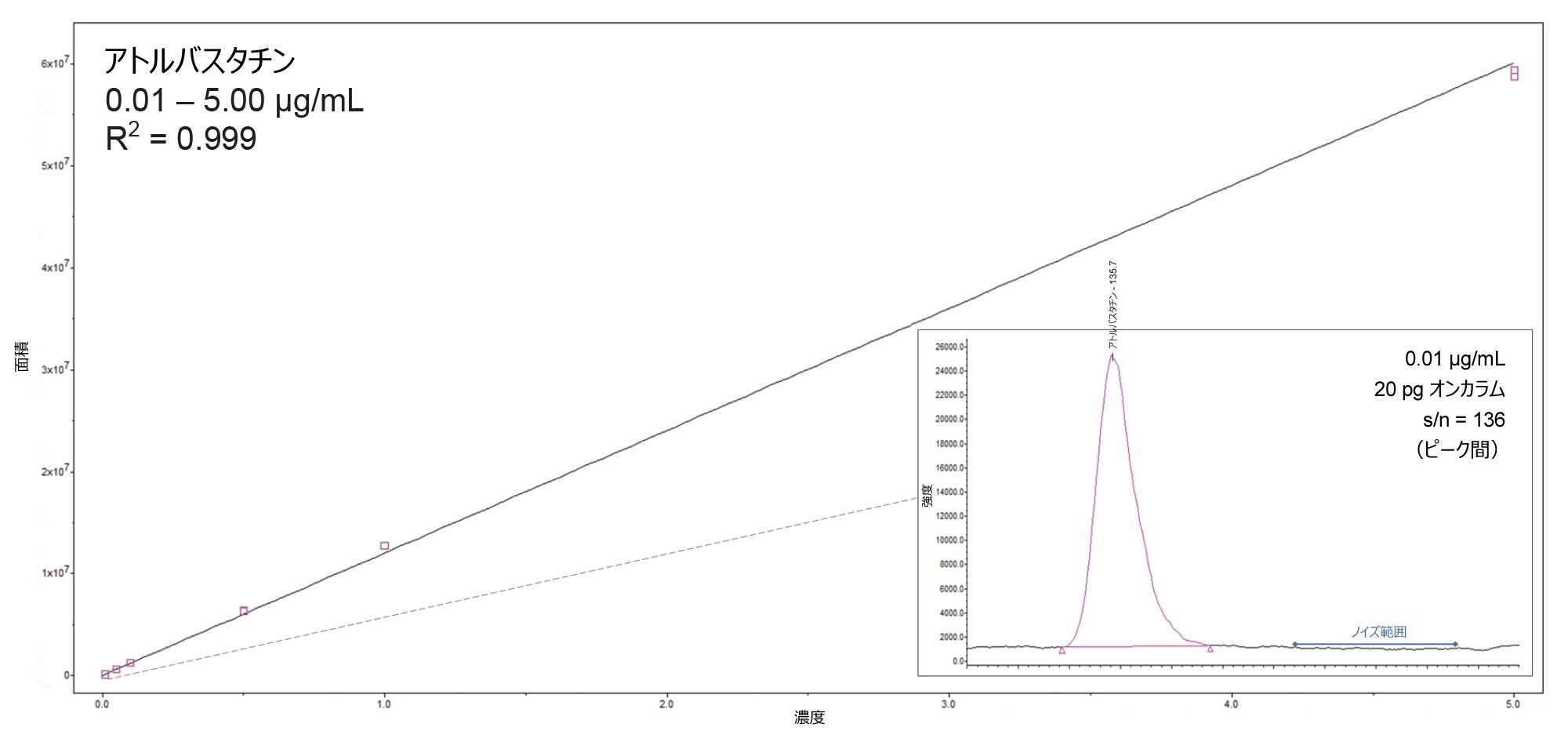
アトルバスタチンのブラケット検量線プロットを図 2 に示します。
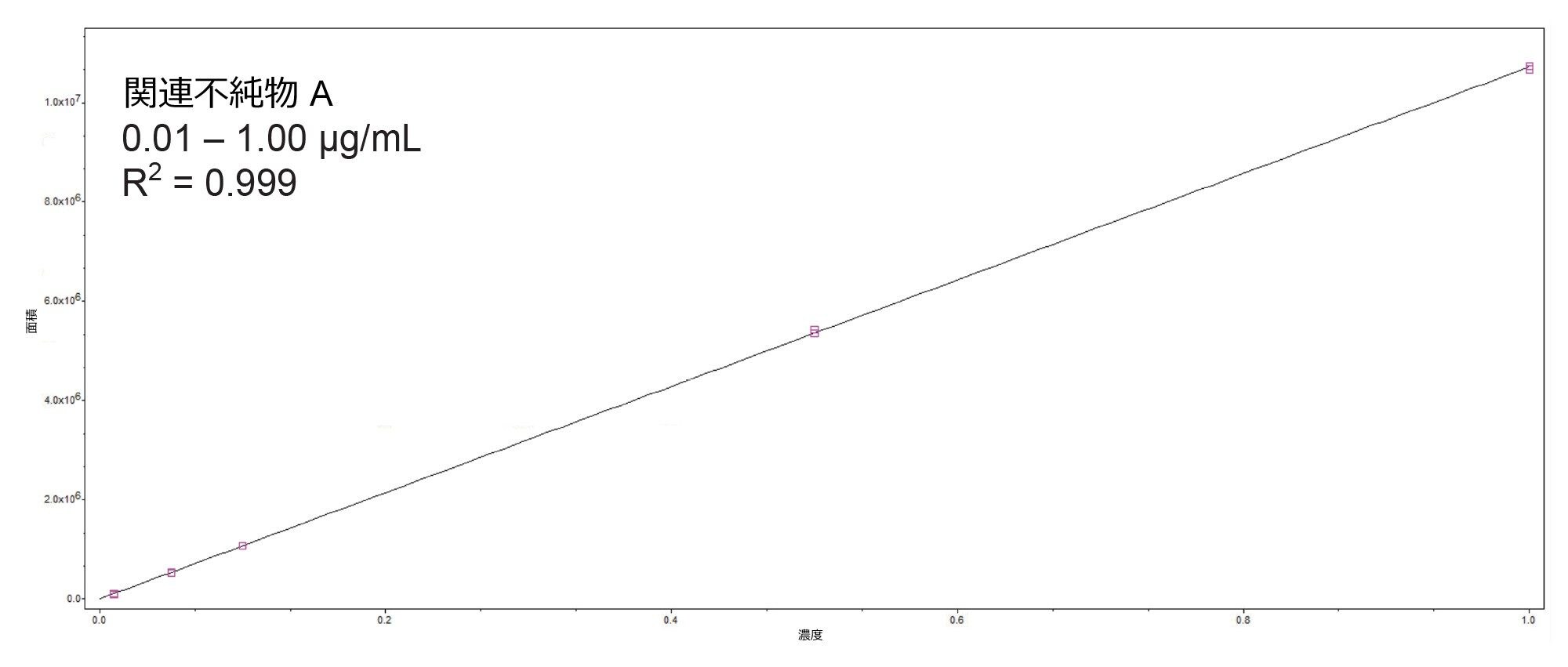
- この化合物は、0.01 ~ 5.0 µg/mL で直線性を示し(1/X 重み付け)、R2 値は 0.999 であることがわかりました
- ブラケット検量線の実測値の割合(% 残差)は、すべての場合で 15.4% 未満でした
- 20 pg オンカラムに相当する最低キャリブレーションポイント(0.01 µg/mL)のピークのシグナル対ノイズ比は 136(ピーク間)でした
一部のアトルバスタチン関連不純物を、直線性および感度についても評価しました(表 1)。すべての場合で R2 値が 0.998 を超え、% 残差の値はすべての場合で 16.3% 未満でした。すべてのキャリブレーションのレスポンスは直線的でした(1/X 重み付け)。
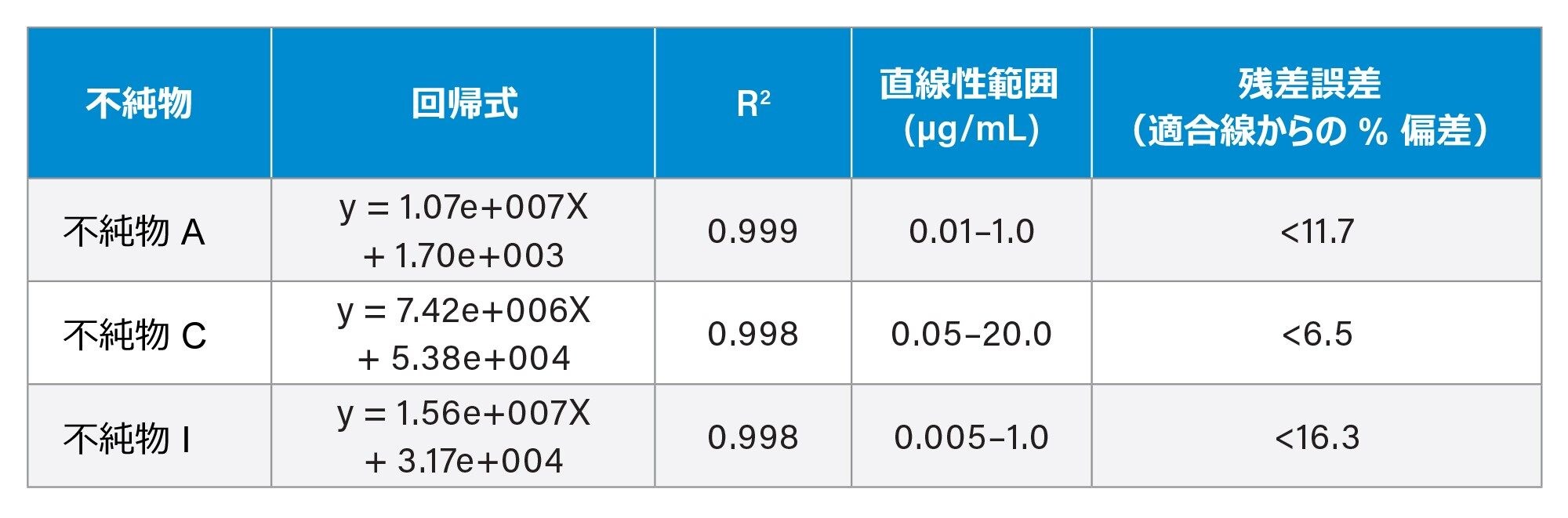
不純物 A の検量線プロットの例を図 3 に示します。高濃度の API を注入したにもかかわらず、この分析法では、分析全体にわたって、後続のブランク注入のいずれにおいてもキャリーオーバーは見られませんでした。この点は、濃度 75 µg/mL までのアトルバスタチン API のすべての注入に該当していました。
0.15% しきい値でのアトルバスタチン不純物の定量
QDa II 質量検出器の感度を関連不純物について評価した後、75 µg/mL のアトルバスタチンサンプルに 0.12% の不純物混合液(A、C、I)を、各不純物が 0.09 µg/mL になるようにスパイクして評価を行いました。便宜上、このサンプルをこれ以降 0.12% しきい値サンプルと呼びます。UV 検出と質量検出の両方を使用して、スキャンモードで化合物を分析しました。このサンプルの UV クロマトグラムおよび関連する MS TIC クロマトグラムの例を図 4 に示します。
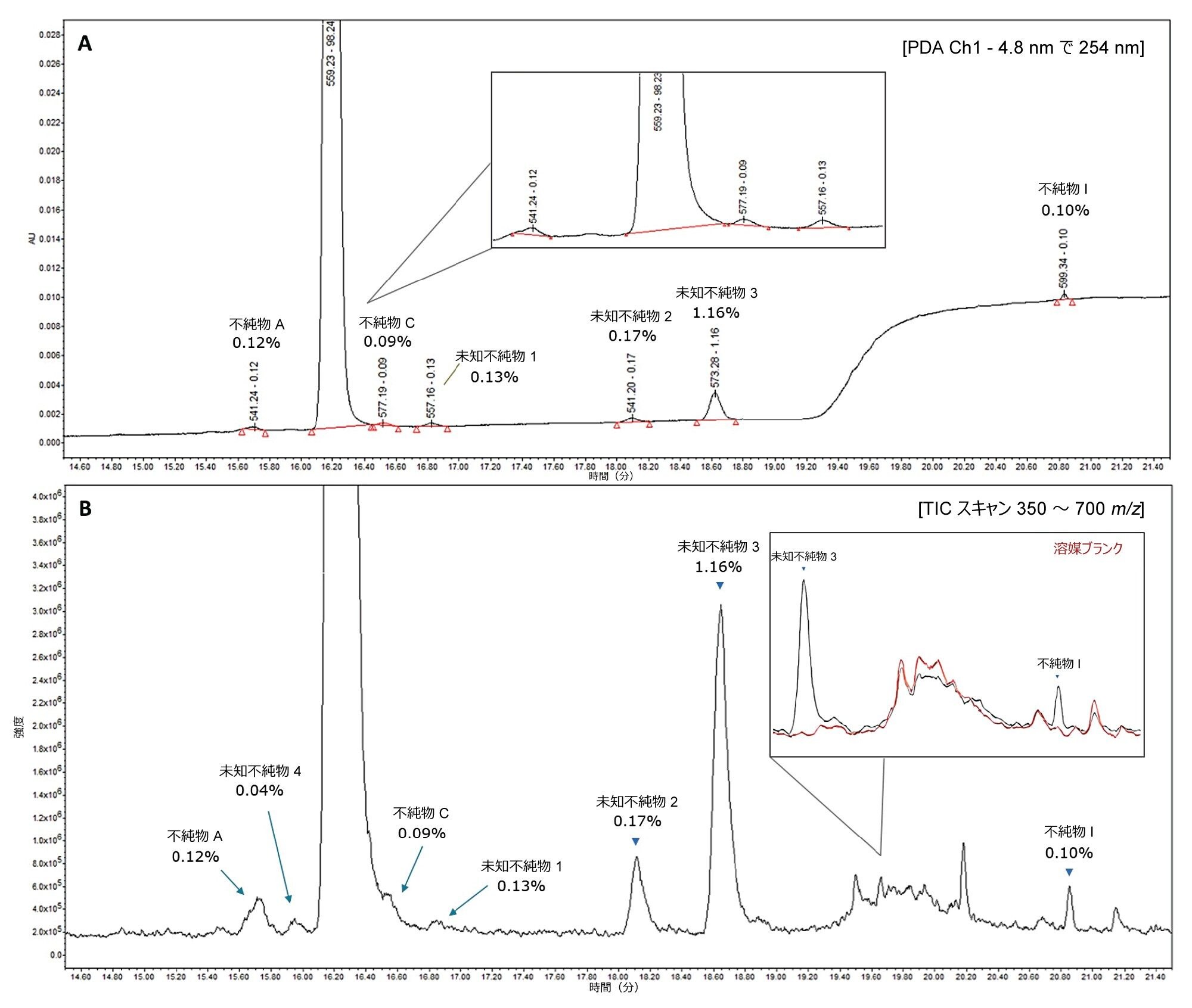
図 4A の PDA トレースでは、3 つの未知ピークがそれぞれ 0.13%、0.17%、1.16% で見られ、これらの相対濃度は図 1 に示した 25 µg/mL サンプルの相対濃度と同等です。図 4B の対応する MS TIC クロマトグラムでは、追加の未知不純物である未知不純物 4 が存在することがより明確にわかります。
図 4B に、同じ分析で取り込んだ溶媒ブランクサンプルの重ね描きも示しています。図 4B の MS TIC クロマトグラム中のラベル付けしていないピークが、API サンプルに固有ではないことがわかります。
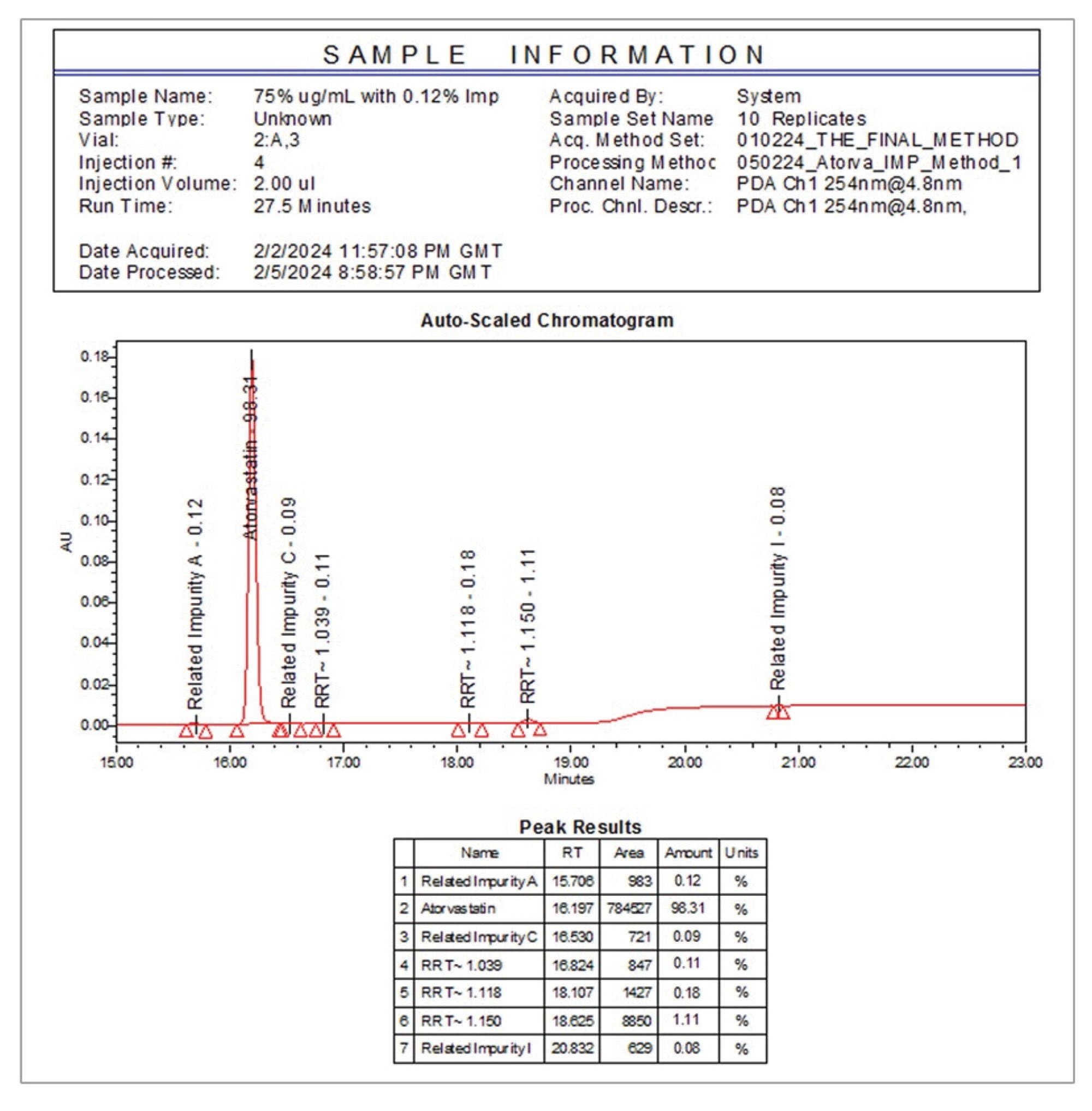
この分析法の再現性を評価するため、0.12% しきい値サンプルを 10 回繰り返し注入しました。この結果を表 2 に示します。各不純物の面積の %RSD は、10 回の注入にわたる %RSD が 13.4% であった関連不純物 A 以外、すべての場合で 9% 未満でした。
![0.12% の不純物 A、C、I をスパイクした 75 µg/mL のアトルバスタチンサンプルの連続 10 回繰り返し注入にわたる、[254 nm、4.8 mm] PDA トレース中の定量ピークの相対 % 面積](/content/dam/waters/ja/app-notes/2024/720008282/720008282en-t2.jpg.82.resize/img.jpg)
不純物 A および C は、記載した時間送液を自動的に廃液に向けるダイバートバルブを使用することで、MS TIC クロマトグラムでより明確に見ることができます。この機能の使用例を図 5 に示します。ここでは、16.08 分 ~ 16.47 分の間(アトルバスタチン API 溶出の RT の間)、送液が転流されています。
![API の溶出時に送液を廃液に向けることにより、不純物 A [m/z 541.1] および不純物 C [m/z 557.1] の定量が容易になるダイバートバルブを使用して得られた MS TIC クロマトグラム](/content/dam/waters/ja/app-notes/2024/720008282/720008282en-f5.jpg.82.resize/img.jpg)
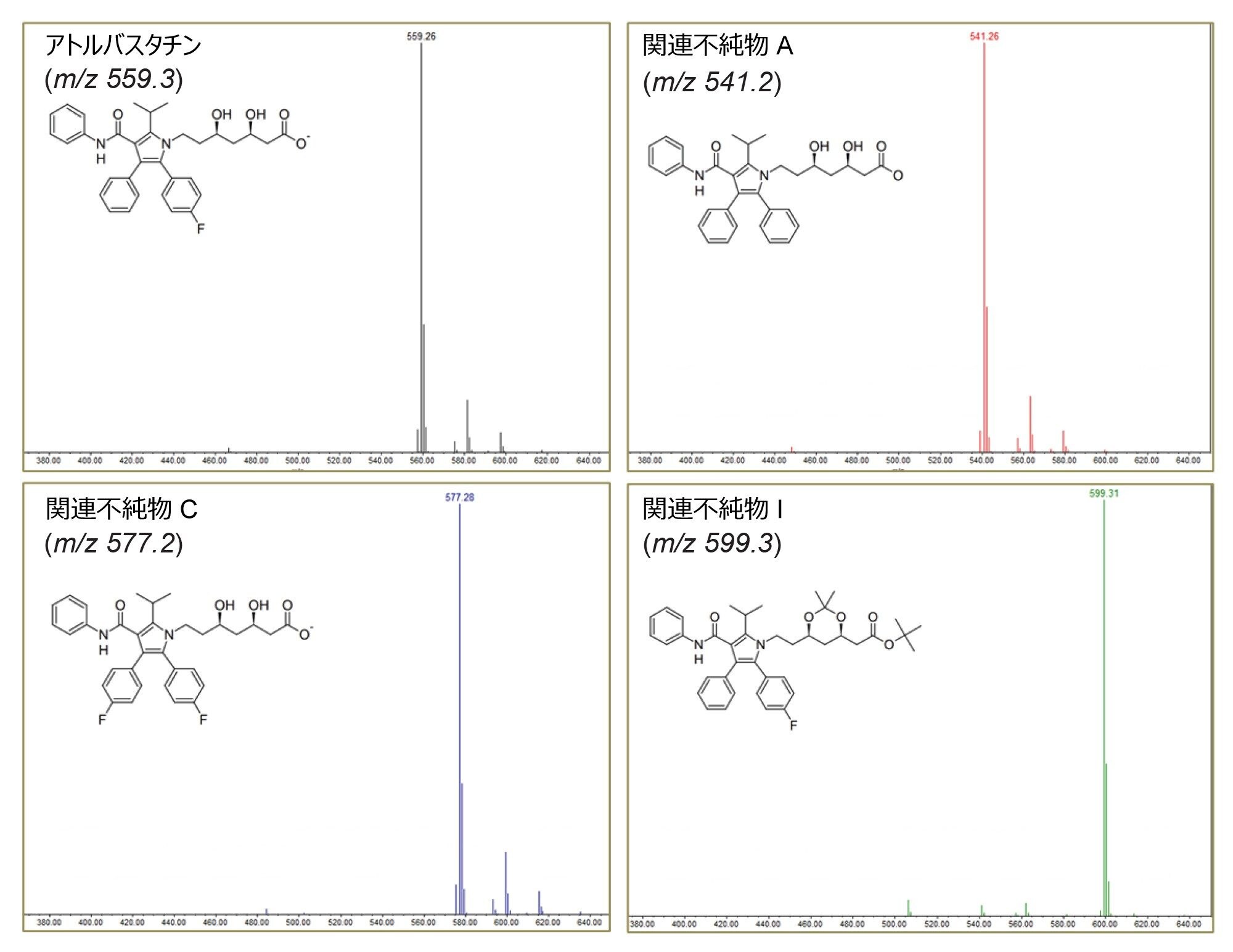
ACQUITY QDa II を使用して、クロマトグラムの各ピークの質量スペクトルを簡単に取得できます(図 6)。
イオン源内フラグメンテーションによる構造情報
調査の結果、未知不純物 1 および 4 の分子量は m/z 557 であり、API の分子量よりわずか 2 Da だけ少ないことが判明しました。文献によると、これはアトルバスタチン化合物の一般的な光分解物であると考えられます6。ただし、未知不純物 2 および 3 は、さらに評価を行い、それぞれの構造をより良く理解する必要があります。
イオン源内フラグメンテーションにより、分析種分子に関する有用な構造情報が得られ、化合物の同定および特性解析に役立つ特徴的なフラグメントイオンの検出が可能になります。
QDa II 質量検出器のコーン電圧を上げることで、イオン源内フラグメンテーションにより、所定の化合物の疑似 MS/MS を行うことができます。この例を図 7 に示します。ここでは、未知不純物 2 と 3 を調査しています。
35 V のコーン電圧を印加すると、未知不純物 3 の m/z 573.3 のプリカーサー質量が m/z 454 にフラグメント化しています(図 7B)。フラグメンテーションパターンから、この化合物は不純物 G またはアトルバスタチンメチルエステルであると考えられます。
この分析では、m/z 611 の付加イオン [M+K]+ が見られ、コーン電圧 50 V でのフラグメンテーションにより、アトルバスタチンメチルエステルの文献評価値7 とよりよく一致する m/z 436、394、376、292 のプロダクトイオンが生じています。ただし、この点を確実にするには、さらなる分析が必要です。
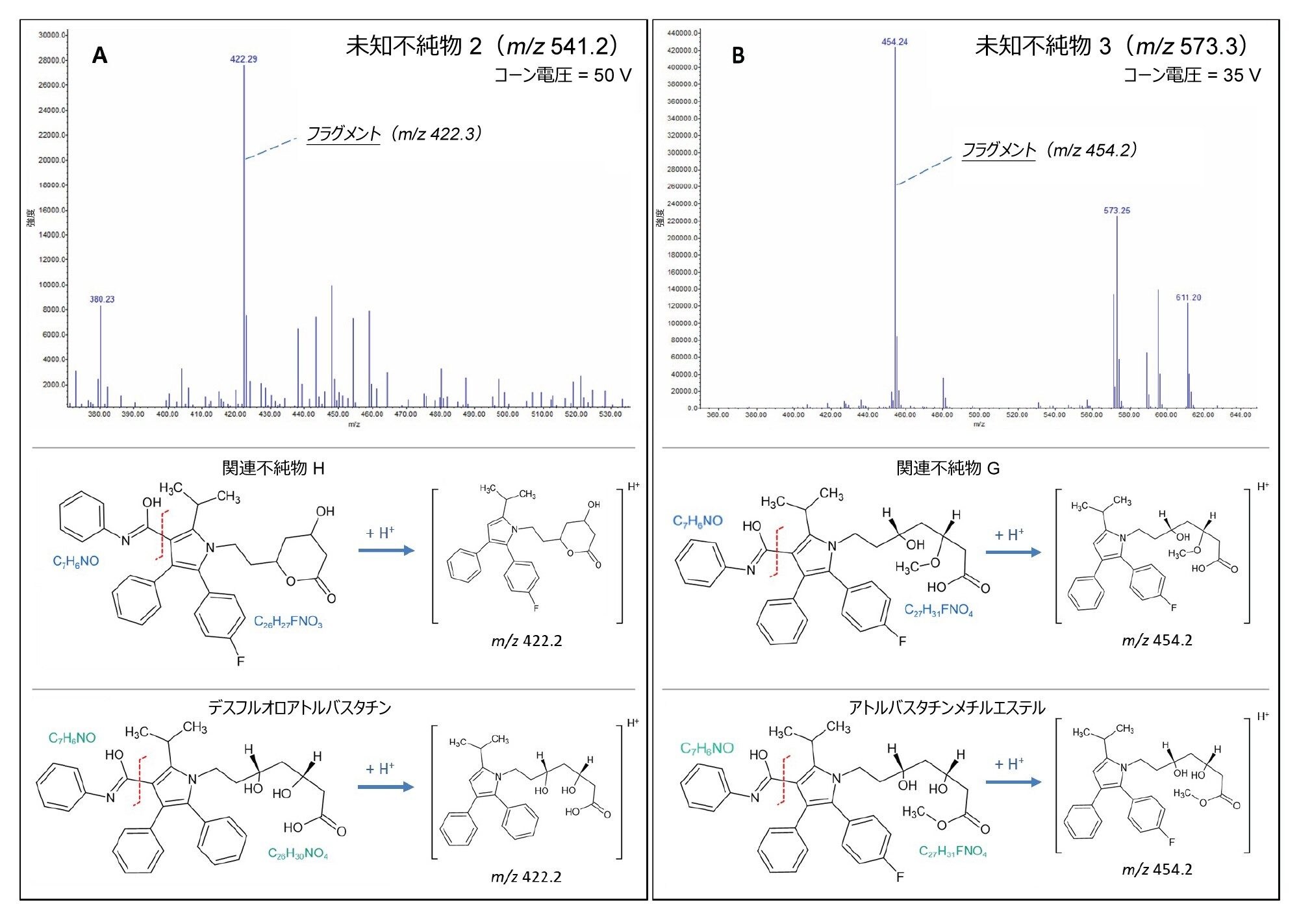
未知不純物 2 の m/z 451.2 プリカーサーイオンに 50V のコーン電圧を印加すると、m/z 422 でフラグメントが生成しました(図 7A)。このフラグメンテーションパターンは、デスフルオロアトルバスタチンと関連不純物 H の両方に一致します。m/z 422 の後続の m/z 380 フラグメントの存在は、この化合物が関連不純物 H である可能性が高いことを示しています。
質量検出による共溶出する不純物の検出
サンプルを 10 ℃ で 2 週間保管して、不純物の経時変化をモニタリングしました。0.12% の不純物混合液を添加した 75 µg/mL のアトルバスタチンサンプルを「分解試験サンプル」に指定しました。図 8 に示す分析により、分解試験サンプル(図 8B)中の関連不純物 A と、API を含まない同様の濃度のサンプル(0.10 µg/mL の不純物混合液)の質量スペクトルの間に興味深い違いがあることが明らかになりました(図 8A)。前者では m/z 557.2 の大幅な増加が観察され、関連不純物 A と共溶出する API の光分解物の存在が示唆されます。
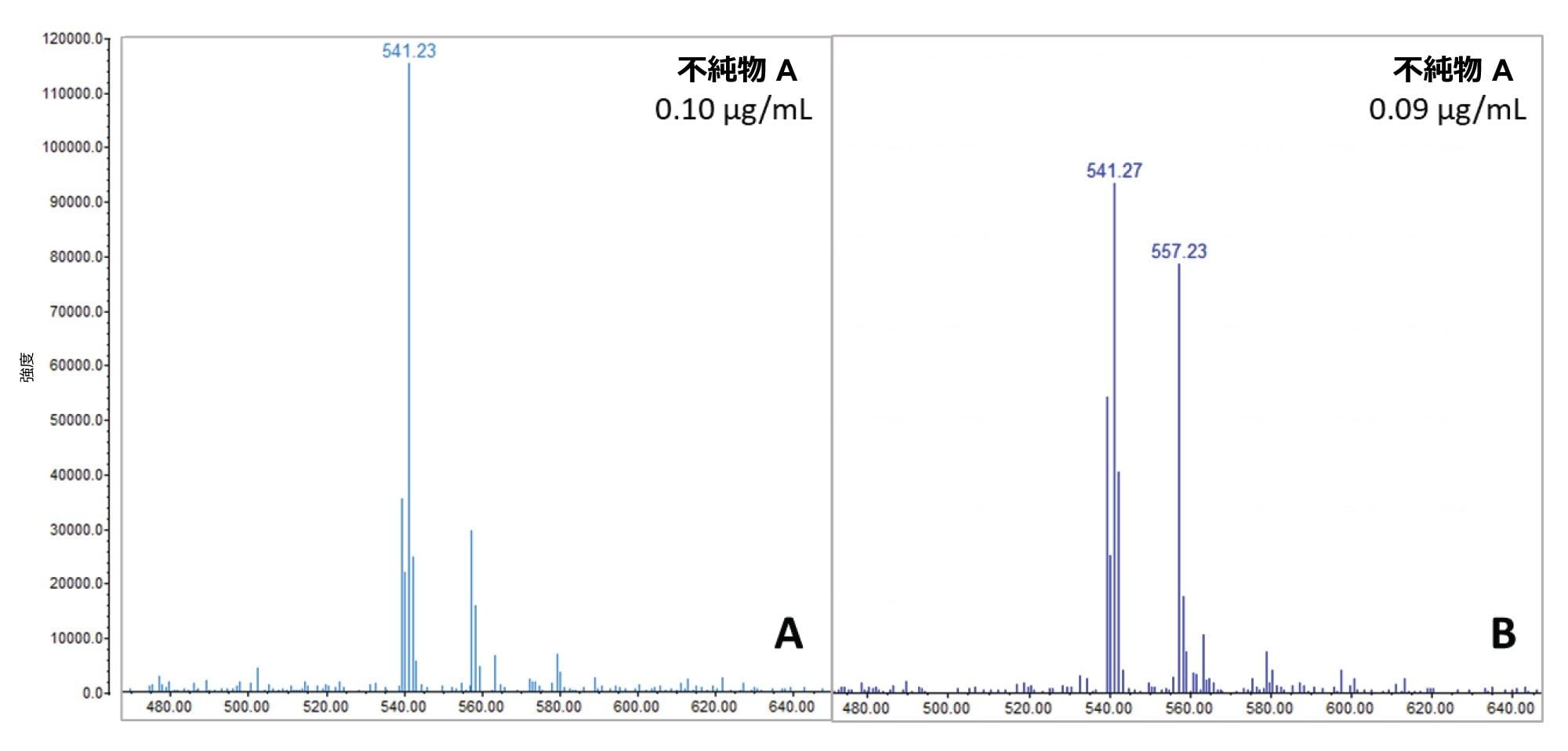
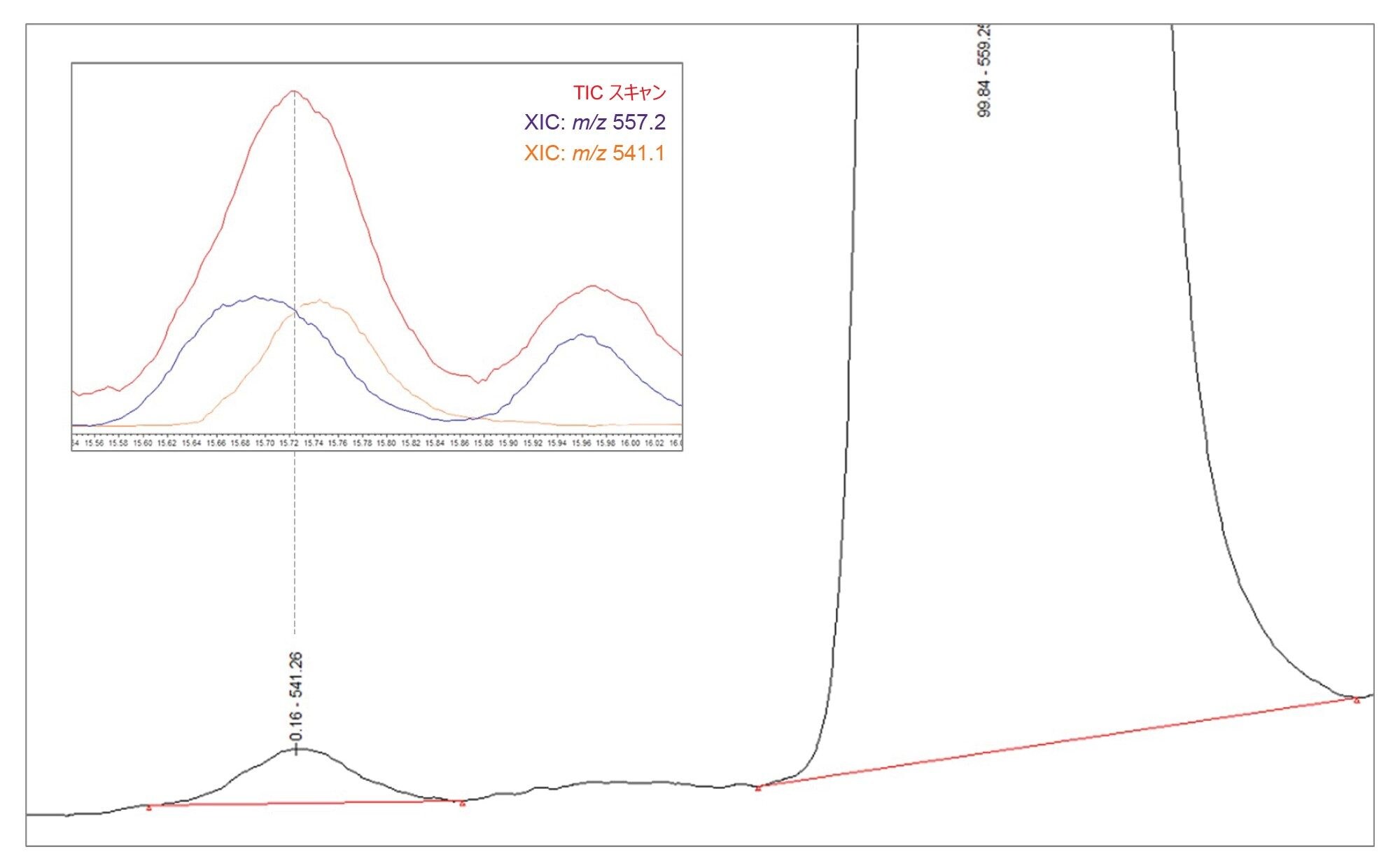
前処理してから約 15 日経過後のサンプルを再定量すると、関連不純物 A の API に対する相対量が、平均 0.10%(表 2 を参照)から 0.16% まで増加しているように見えました(図 9)。この違いは、関連不純物 A の濃度の変化によるものではなく、別の化合物との共溶出によるものであり、API が分解するにつれてより顕著になっています。このため、UV クロマトグラフィーを使用した定量では、過大評価になってしまいます。共溶出を図 9 に示します。ここでは、関連不純物 A(m/z 541.1)の XIC が、m/z 557.2 の不純物の XIC および MS TIC スキャンに重ね描きされています。このことから、不純物の正確な定量と共溶出の適切な管理を確実に行うための質量検出の重要性が浮き彫りになっています。
データレポート作成の強化
このアプリケーションでは、Empower CDS のレポート機能を存分に活用することにより、サンプルバッチ内の不純物の相対濃度の合理的な評価が容易に行えます(図 10 参照)。この頑健な機能により、ユーザーは、十分な情報に基づいた迅速な決定を行うことができ、ワークフローのコンプライアンスとデータインテグリティの両方が確実に得られます。
結論
この研究では、以下の機能を実証することにより、API 中の不純物のルーチン試験における既存の UV ワークフローに ACQUITY QDa II 質量検出器を採用することが、エンドユーザーにとって大きなメリットとなることを実証しています。
- ピークに分子量の注釈が簡単に付けられるため、分析内での質量確認がシンプルになり、既知の不純物の確認プロセスが合理化されます
- 不純物のレベルを精密に定量して、アトルバスタチン関連不純物 A、C、I を、API に対して相対的に 0.15% 未満という適格性評価しきい値に維持することができます
- イオン源内フラグメンテーションにより構造的洞察が強化され、未知不純物の分子量の確認が容易になり、化合物の構造に対する理解がより深まります
- 質量検出から得られる有益な情報により、既知の不純物の報告におけるバイアスを防ぎ、干渉物の検出が可能になるとともに、製品品質を維持し、製品リリースまでの時間を短縮できます
この分析アプローチを Empower CDS ソフトウェアと組み合わせることで、不純物ワークフローが向上し、シンプルかつ高い効率性が実現して、コンプライアンスおよびデータインテグリティの基準を維持することができます。
参考文献
- Vukkum P, Moses Babu J, Muralikrishna R. Stress Degradation Behavior of Atorvastatin Calcium and Development of a Suitable Stability-Indicating LC Method for the Determination of Atorvastatin, its Related Impurities, and its Degradation Products.Sci Pharm.Jan-Mar;81(1):93–114.2013.
- Impurities in EW drug substances Q3A(R2)–ICH (Oct 2006) ICH–Q3 Guidelines.Available at: https://database.ich.org/sites/default/files/Q3A%28R2%29%20Guideline.pdf (Accessed: 09 February 2024).
- European Pharmacopoeia (Ph.Eur.)11th Edition.Available at: https://www.edqm.eu/en/european-pharmacopoeia-ph.-eur.-11th-edition (Accessed: 09 February 2024).
- Shulyak, N. et al. ‘Development of a Novel, Fast, Simple HPLC Method for Determination of Atorvastatin and Its Impurities in Tablets’, Scientia Pharmaceutica, 89(2), p.16. 2021.
- Piponski, M. et al.‘Concepts in Development of Fast, Simple, Stability Indicating HPLC Method for Analysis of Atorvastatin Related Compounds in Tablets’, Journal of Analytical & Pharmaceutical Research, 7(4), pp.450–457.2018.
- Stach, J. et al. ‘Synthesis of Some Impurities and/or Degradation Products of Atorvastatin’, Collection of Czechoslovak Chemical Communications, 73(2), pp.229–246.2008.
- Mornar, A., Damić, M. and Nigović, B. ‘Separation, Characterization, and Quantification of Atorvastatin and Related Impurities by Liquid Chromatography-Electrospray Ionization Mass Spectrometry’, Analytical Letters, 43(18), pp.2859–2871.2010.
720008282JA、2024 年 4 月