臨床研究のための血漿中抗うつ薬の分析
研究目的のみに使用してください。診断用には使用できません。
要約
抗うつ薬およびその他のクラスの神経薬理薬の薬物動態および薬物相互作用が報告されています1。 したがって、信頼性の高い分析法が、臨床研究で重要な役割を果たします。
本研究では、内部標準を含む血漿の除タンパクを使用して、抗うつ薬を分析する臨床研究の分析法について説明します。Waters ACQUITY™ UPLC™ I-Class システムで Waters XSelect™ Premier HSS T3 カラムに続いて Xevo™ TQD 質量分析計を接続して検出を行ったところ、クロマトグラフィーでの溶出が 5 分以内に完了していました(図 1)。

アプリケーションのメリット
- クロマトグラフィーおよび質量検出器によって分析の選択性を実現
- 少量のサンプルを使用したシンプルで低コストのサンプル前処理
- 分析時間の短縮
実験方法
サンプル前処理
血漿キャリブレーターと品質管理物質は、BioIVT(英国、ウェストサセックス)から供給されたプールしたヒト血漿を使用して社内で調製しました。濃縮ストック溶液は、Cambridge Bioscience(英国、ケンブリッジシャー)、Merck Life Science(英国、ドーセット)、Toronto Research Chemicals(カナダ、オンタリオ)から供給された認証済み粉末から調製しました。安定同位体標識内部標準は、Cambridge Bioscience(英国、ケンブリッジシャー)、Merck Life Science(英国、ドーセット)、Toronto Research Chemicals(カナダ、オンタリオ)から提供を受けました。キャリブレーション範囲は、ミルタザピン(5 ~ 500 ng/mL)およびトラゾドン(30 ~ 3000 ng/mL)を除くすべての分析種で 10 ~ 1000 ng/mL でした。
サンプル抽出
微量遠心チューブ内の 50 µL のサンプルに、150 µL の内部標準のアセトニトリル溶液を加えました。内部標準の濃度は表 1 に示します。
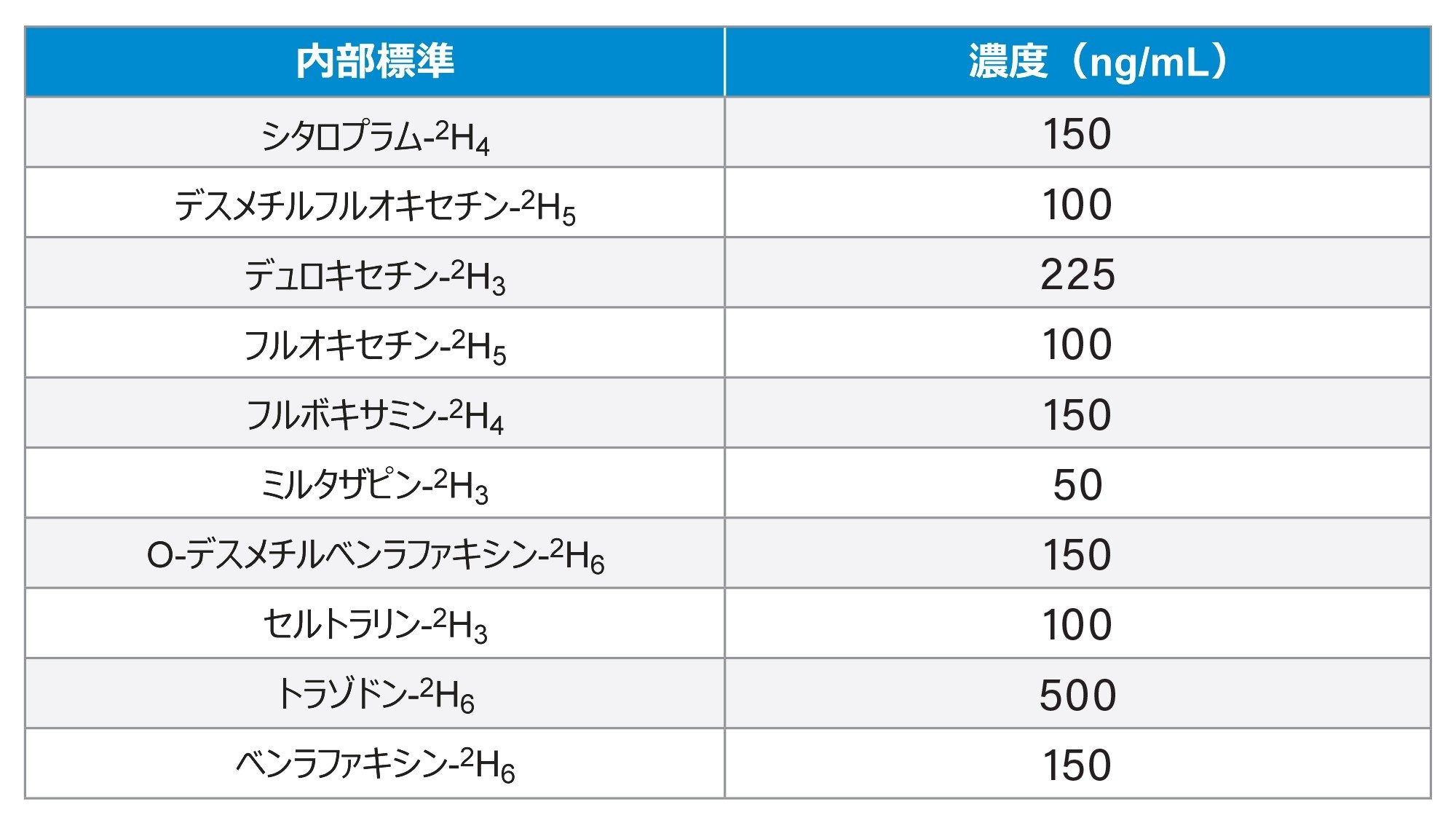
マルチチューブボルテックスミキサーにチューブを配置し、2,500 rpm で 30 秒間ボルテックス混合してから、16,100 g で 2 分間遠心分離しました。上清 50 µL を 1 mL の 96 ウェルプレートに移し、水を 450 µL 加えました。
移動相とカラムは、他の薬剤(抗てんかん薬、三環系抗うつ薬、抗精神病薬など)との適合性を考慮して選択しました。
UPLC 条件
|
システム: |
ACQUITY UPLC I-Class、FTN 搭載 |
|
ニードル: |
30 μL |
|
カラム: |
XSelect HSS T3 カラム、2.5 µm、2.1 × 100 mm(製品番号:186009831) |
|
移動相 A: |
水 + 2 mM 酢酸アンモニウム |
|
移動相 B: |
メタノール+ 2 mM 酢酸アンモニウム |
|
ニードル洗浄溶媒: |
80% メタノール水溶液 + 0.1% ギ酸 |
|
パージ溶媒: |
水:メタノール(40:60、v/v) |
|
シール洗浄溶媒: |
20% メタノール水溶液 |
|
カラム温度: |
45 °C(プレカラムヒーターアクティブ) |
|
注入量: |
20 μL |
グラジエントテーブル
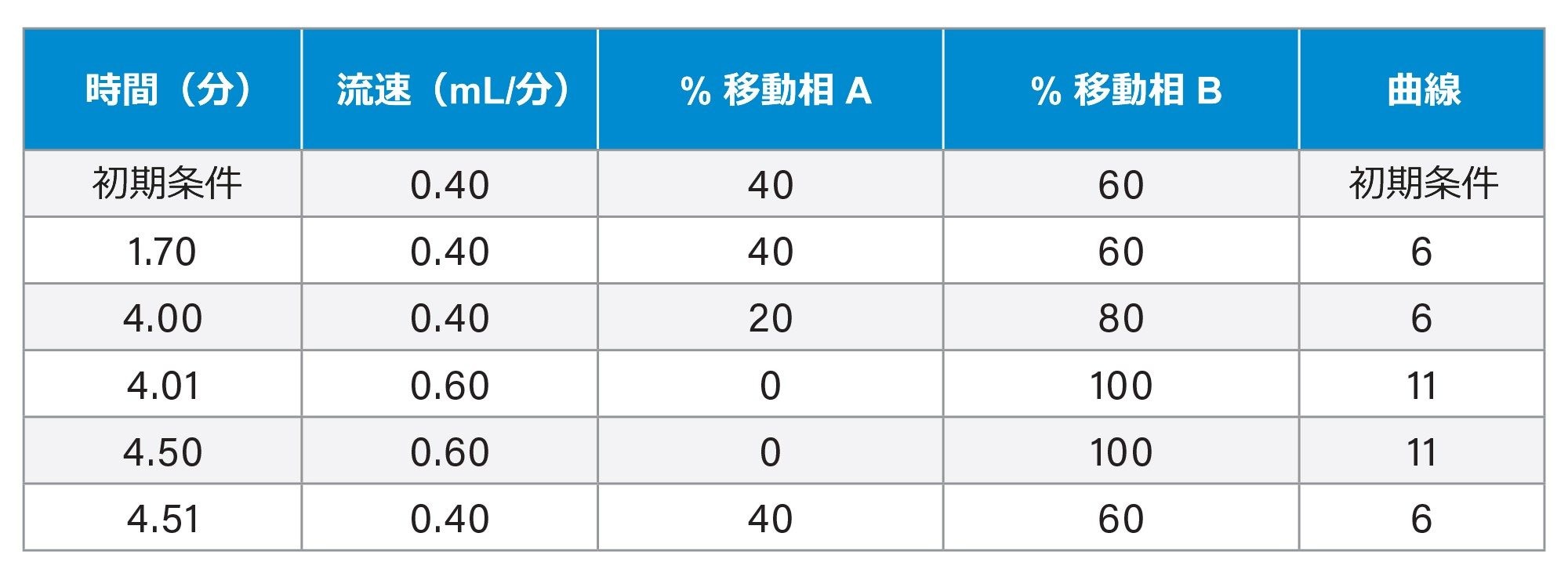
|
分析時間: |
5.0 分(注入間時間 5.7 分) |
MS 条件
|
システム: |
Xevo TQD |
|
分解能: |
MS1(0.7 FWHM)、MS2(0.7 FWHM) |
|
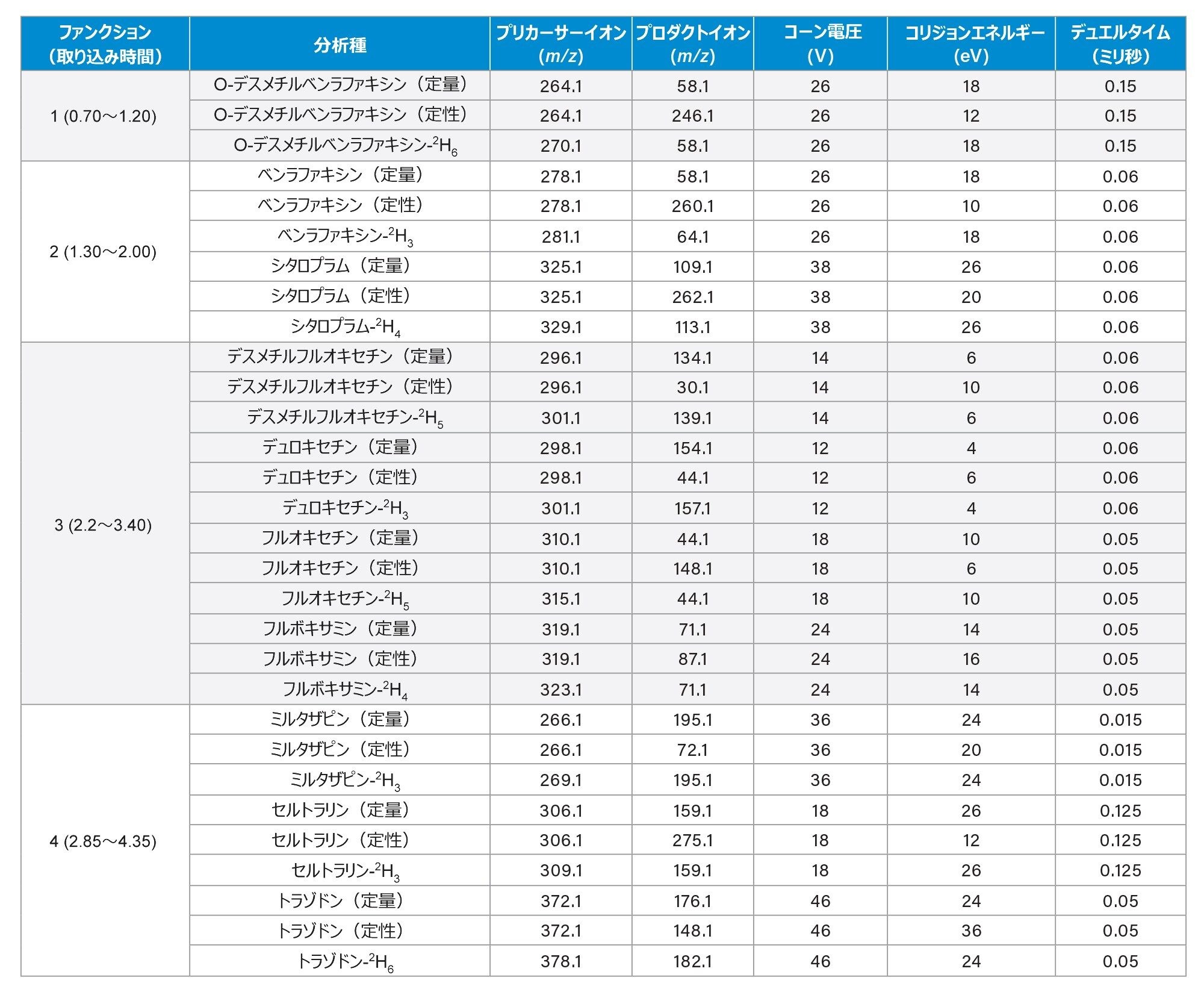
取り込みモード: |
マルチプルリアクションモニタリング(MRM)(詳細については、表 2 を参照してください |
|
極性: |
ESI+ イオン化 |
|
キャピラリー電圧: |
0.5 kV |
|
イオン源温度: |
150 ℃ |
|
脱溶媒温度: |
400 ℃ |
|
コーンガス: |
50 L/時間 |
|
脱溶媒ガス流量: |
1,000 L/時間 |
|
スキャン間遅延: |
0.003 秒 |
|
極性/モード切り替えのスキャン間遅延: |
0.020 秒 |
|
チャンネル間遅延: |
0.020 秒 |
データ管理
MassLynx™ v4.2(TargetLynx™ XS アプリケーションマネージャー搭載)
結果および考察
図 2 に、クロマトグラムの例を示します。

1,000 ng/mL のシタロプラム、デスメチルフルオキセチン、デュロキセチン、フルオキセチン、フルボキサミン、O-デスメチルベンラファキシン、セルトラリン、ベンラファシン、500 ng/mL のミルタザピンおよび 3,000 ng/mL のトラゾドンを含む血漿サンプルの分析後、システムキャリーオーバーは観察されませんでした。
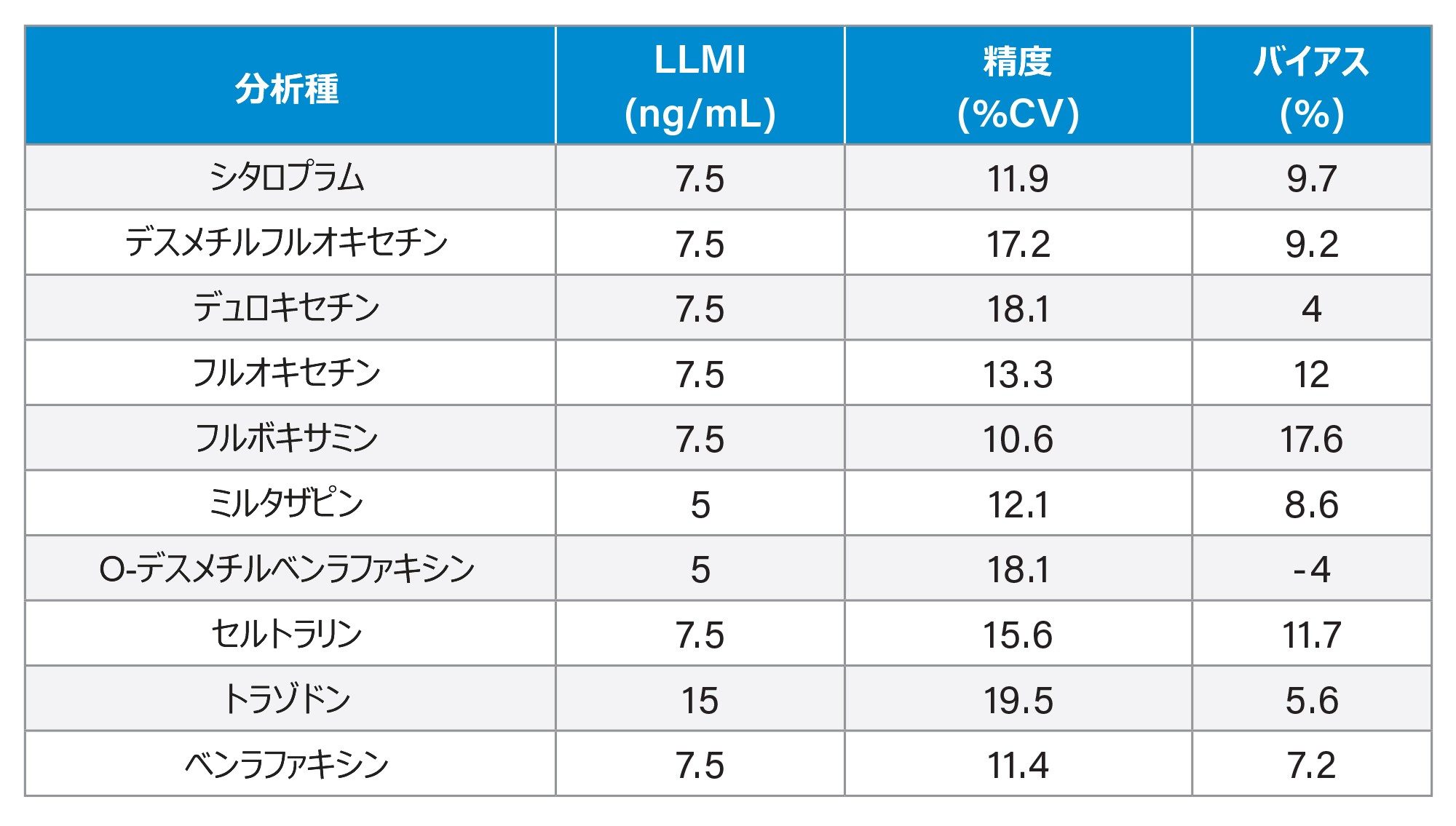
5 日間にわたって血漿中に調製した低濃度サンプルを 10 回繰り返し抽出・定量することで、分析感度を評価しました。調査の結果、この分析法では、表 4 に示す濃度で正確な定量(≤ 20% CV、≤15% バイアス)が可能であることが示されました。
唯一の例外はフルボキサミンで、バイアスが 15% をわずかに超えていましたが、17.6% では 20% 以下で、10 ng/mL のキャリブレーター 1 の許容可能な % 偏差です。
合計精度は、連続しない 5 日間にわたって 3 濃度の血漿プールを 5 回繰り返しで抽出・定量することによって決定しました(n = 25)。各レベルで QC 試料を 5 回繰り返し分析することによって併行精度を評価しました。図 3 にこれらの実験の結果を示します。濃度が 12.5、37.5、400 ng/mL のミルタザピン、および 75、225、2,400 ng/mL のトラゾドンを除くすべての分析種で、評価した 3 濃度(25、75、800 ng/mL)すべてで合計精度と併行精度が 10.0% CV 以下でした。
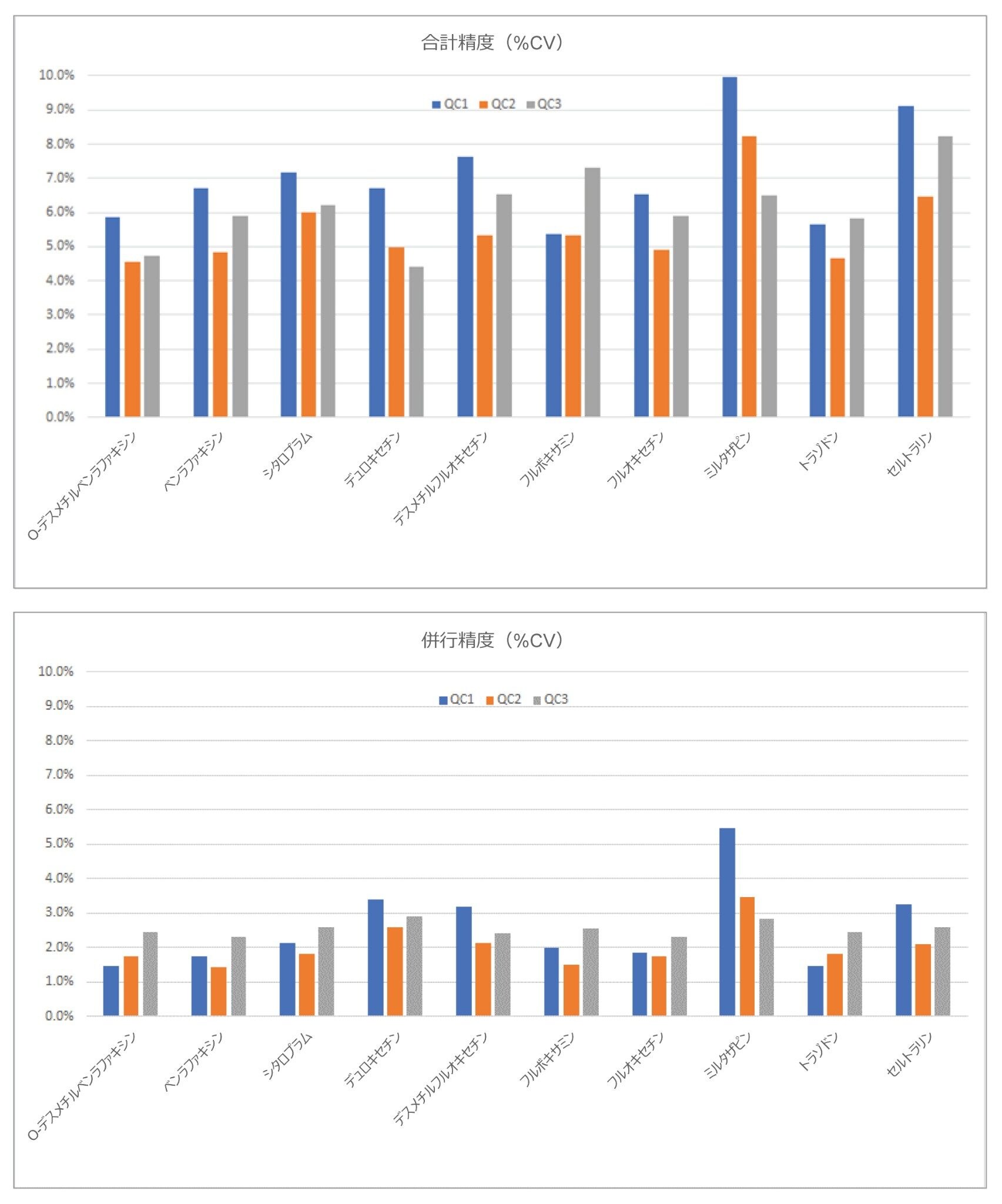
この分析法は、低濃度のプールと高濃度のプールを一定の範囲にわたって既知の比率で混合した場合、O-デスメチルベンラファキシンおよびデュロキセチンについて 7.7 ~ 1,300 ng/mL の範囲にわたって直線性があることが分かりました。シタロプラム、デスメチルフルオキセチン、フルボキセチン、フルボキサミン、セルトラリン、ベンラファクシンはすべて 7.7 ~ 1,300 ng/mL の範囲で二次近似を示すと判定され、同様に、ミルタザピンは 3.8 ~ 650 ng/mL、トラゾドンは 23 ~ 3,900 ng/mL で二次近似を示すと考えられました。
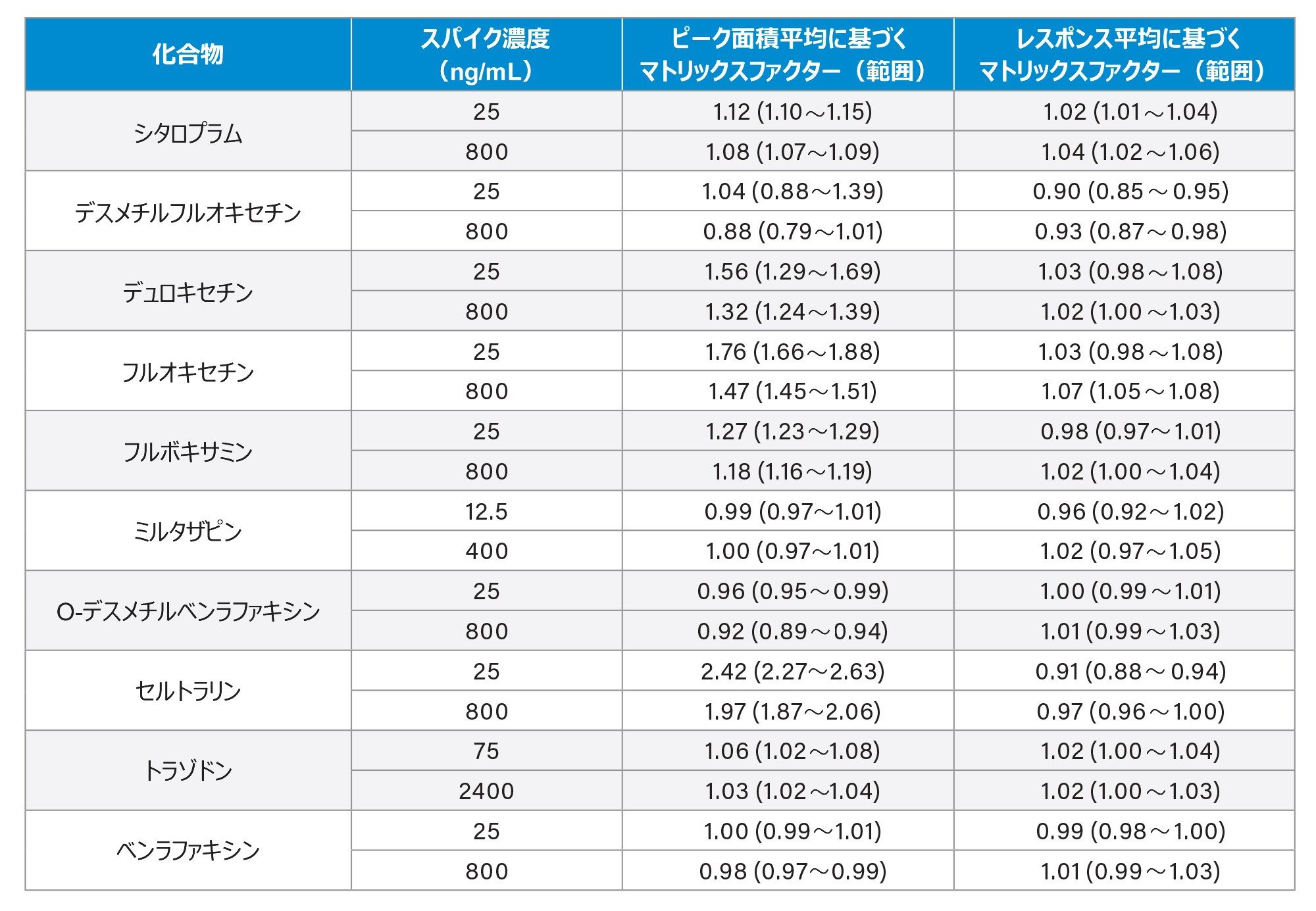
マトリックス効果は、血漿中の低 QC 濃度(QC1)および高 QC 濃度(QC3)(n = 6)で、同等の濃度になるようにスパイクした抽出溶媒サンプルの割合として評価しました。分析種対内部標準のレスポンス比を用いる計算を、内部標準によるシグナルの増強または抑制の補正の指標としました(表 5)。
高濃度でスパイクした内因性化合物(アルブミン、ビリルビン、コレステロール、クレアチニン、トリグリセリド、尿酸)からの潜在的な干渉を、低濃度および高濃度のプールした血漿サンプル(QC1 濃度およびQC3 濃度)からの回収率を決定することによって評価しました(n=3)。回収率は 86.9 ~ 112.9% の範囲でした。回収率の範囲が 85% ~ 115% を超える場合、干渉物質があると見なしました。さらに、O-デスメチルベンラファキシンの同重体トラマドールからの完全なクロマトグラフィー分離も確立されました。
結論
この分析法では、3 桁にわたる 10 種類の抗うつ薬およびその代謝物について、わずか 50 µL のサンプルを使用する迅速で安価なサンプル前処理により、5 分の分析時間で 10.0 % CV 未満の合計精度と併行精度が得られることが実証されました。
さらに、システムのキャリーオーバーは試験範囲内では見られませんでした。マトリックス効果が存在する場合は、安定同位体標識内部標準を使用して、非常に効果的に補正されました。
720007859JA、2023 年 2 月