法中毒学における尿中の疼痛管理薬および乱用薬物の UPLC-MS/MS 分析のための簡単な直接希釈注入メソッド
法中毒学目的のみに使用してください。
要約
このアプリケーションブリーフでは、法中毒学における尿中の疼痛管理薬および乱用薬物の UPLC-MS/MS 分析のための簡単な直接希釈注入メソッドについて説明し、ウォーターズアプリケーションノート 720006187JA に記載されている方法の代替となるアプローチを紹介します。サンプル前処理を、固相抽出(SPE)から単一の希釈ステップに簡素化しても、キャリーオーバーが最小限に抑えられます。また、一貫したマトリックス効果および正確な定量データが得られて、パネル内の大部分の分析種に必要な分析感度が得られます。より高い分析感度が必要な化合物については、元のアプリケーションノートの記載に従って、さらなるサンプルクリーンアップを行うことを推奨します。
ACQUITY™ UPLC™ BEH™ C18 カラムを使用することにより、大規模な化合物のパネルを迅速に分析できると同時に、同重体化合物からの干渉を回避して必要な分離をすべて確実に維持できます。
Waters™ Xevo™ TQ-S micro IVD により、広いダイナミックレンジにわたる分析種の大規模なパネルを正確に定量し、2 ng/mL の分析種も 2500 ng/mL の分析種も同時に定量することができます。

サンプル抽出、クロマトグラフィー分離、MS/MS 検出の組み合わせにより、簡単なワークフローおよび迅速かつ精密な分析法が得られます。この分析法は、Hamilton STAR または STARlet システムを用いて自動化することもできます。
アプリケーションのメリット
- 指定薬物の包括的なパネルを分析するための迅速かつ簡単な分析法
- 簡単な直接希釈注入によるサンプル抽出メソッド
- 最小限のキャリーオーバーと一貫したマトリックス効果および回収率
- 指定薬物の大規模なパネルの精密な定量
はじめに
法中毒学の分析に使用される分析種のパネルには通常、違法薬物や一般的な乱用薬物が含まれています。複数の薬物クラスを包括的に把握するには、多くの場合、複数の分析法が使用されます。これらの分析法には、免疫測定法、GC-MS、LC-MS/MS、またはこれらの分析法の組み合わせが含まれます。ウォーターズでは、法中毒学における明確な同定に適した分析感度、選択性、正確性を達成するために、包括的な薬物のパネルを定量できる分析法を開発しました。
この分析法では、直接希釈注入アプローチを使用した簡単なサンプル抽出手順と、ACQUITY UPLC BEH C18 カラムを使用した迅速で再現性のあるクロマトグラフィー分析法を組み合わせることにより、干渉する可能性のある分析種のクリティカルペアすべてについてベースライン分離が実現されています。拡張ダイナミックレンジ(XDR)を備えた Waters Xevo TQ-S micro IVD 質量分析計により、この多様な化合物群に必要とされる分析感度とダイナミックレンジ機能が得られました。この分析法では、疼痛管理薬および乱用薬物について適切な結果が得られることが示されていますが、簡素化した直接希釈注入ワークフローを使用するため、分析感度に関しては若干の制限があります。これらの少数の分析種に低濃度までの分析感度が必要な場合は、ウォーターズアプリケーションノート 720006187JA に記載されているように、さらなるサンプルクリーンアップが推奨されます。
実験方法
サンプル抽出
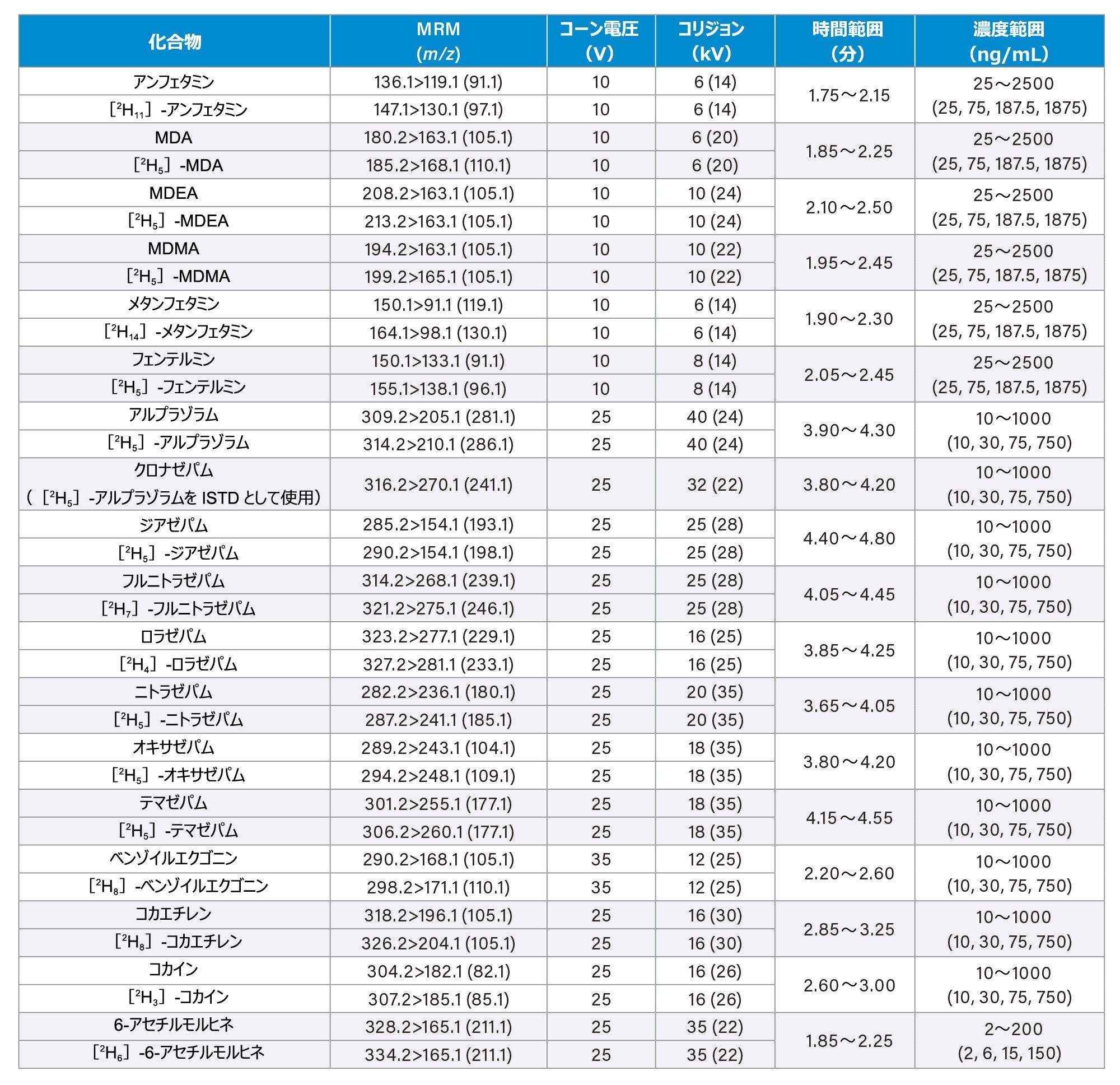
すべての標準試料は、Cerilliant(Merck Life Sciences、英国、ギリンガム)、Toronto Research Chemicals(オンタリオ州ノースヨーク)、および Cambridge Bio Sciences UK(英国、ケンブリッジ)から入手しました。分析種に応じて、メタノールで濃度 2、10、25 µg/mL のストック混合液を調製しました。内部標準作業溶液は、50/50(v/v)メタノール/水で 100 ng/mL の濃度に調製しました。安定同位体標識内部標準が容易に入手できないクロナゼパム、デヒドロノルケタミン、メテドロン、ノロキシモルフォン、α-ピロリジノバレロフェノン(α-PVP)代謝物 1 を除き、すべての化合物に安定同位体標識内部標準を使用しました。標準試料は、ストック溶液を希釈した後、プールしたブランク尿に希釈液をスパイクして調製しました。品質管理(QC)サンプルも、ストック溶液を希釈した後、プールしたブランク尿に希釈液をスパイクして作成しました。すべての分析種について、保持時間とキャリブレーション範囲を、付録 1 に記載しています。
Hamilton リキッドハンドリングロボットを活用することにより、サンプル抽出を半自動化して、サンプルチューブのバーコードから解析済みサンプルまでのサンプル追跡が可能になりました。
25 µL の尿サンプルをウォーターズの 700 µL 丸型 96 ウェルコレクションプレートに移し、内部標準作業溶液を添加して完全に混合します。サンプルは蒸留水/ギ酸溶液で希釈し、混合してから UPLC-MS/MS システムに注入します。
LC 条件
|
LC システム: |
ACQUITY UPLC I-Class FL IVD |
|
カラム: |
ACQUITY UPLC BEH C18、1.7 µm、2.1 × 100 mm |
|
カラム温度: |
40 ± 2 ℃ アラーム |
|
注入量: |
20 µL |
|
移動相 A: |
0.1% ギ酸水溶液 |
|
移動相 B: |
0.1% ギ酸アセトニトリル溶液 |
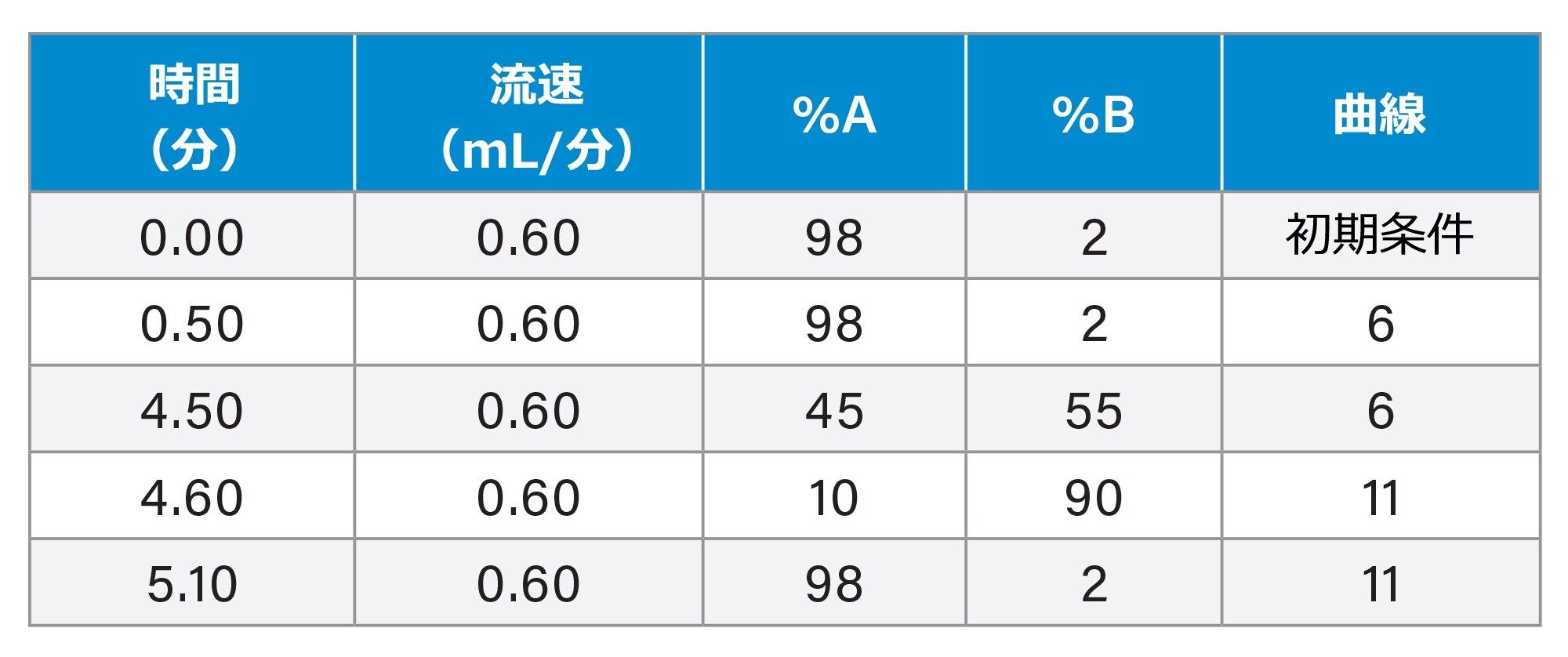
グラジエントテーブル
MS 条件
|
MS システム: |
Xevo TQ-S microIVD |
|
イオン化モード: |
ESI+ |
|
キャピラリー電圧: |
0.8 kV |
コーン電圧、コリジョンエネルギー、マルチプルリアクションモニタリング(MRM)トランジションなどの MS メソッドのパラメーターは、付録 1 に記載されています。
データ管理
|
MS ソフトウェア: |
MassLynx |
|
インフォマティクス: |
TargetLynx™ |
結果および考察
クロマトグラフィー
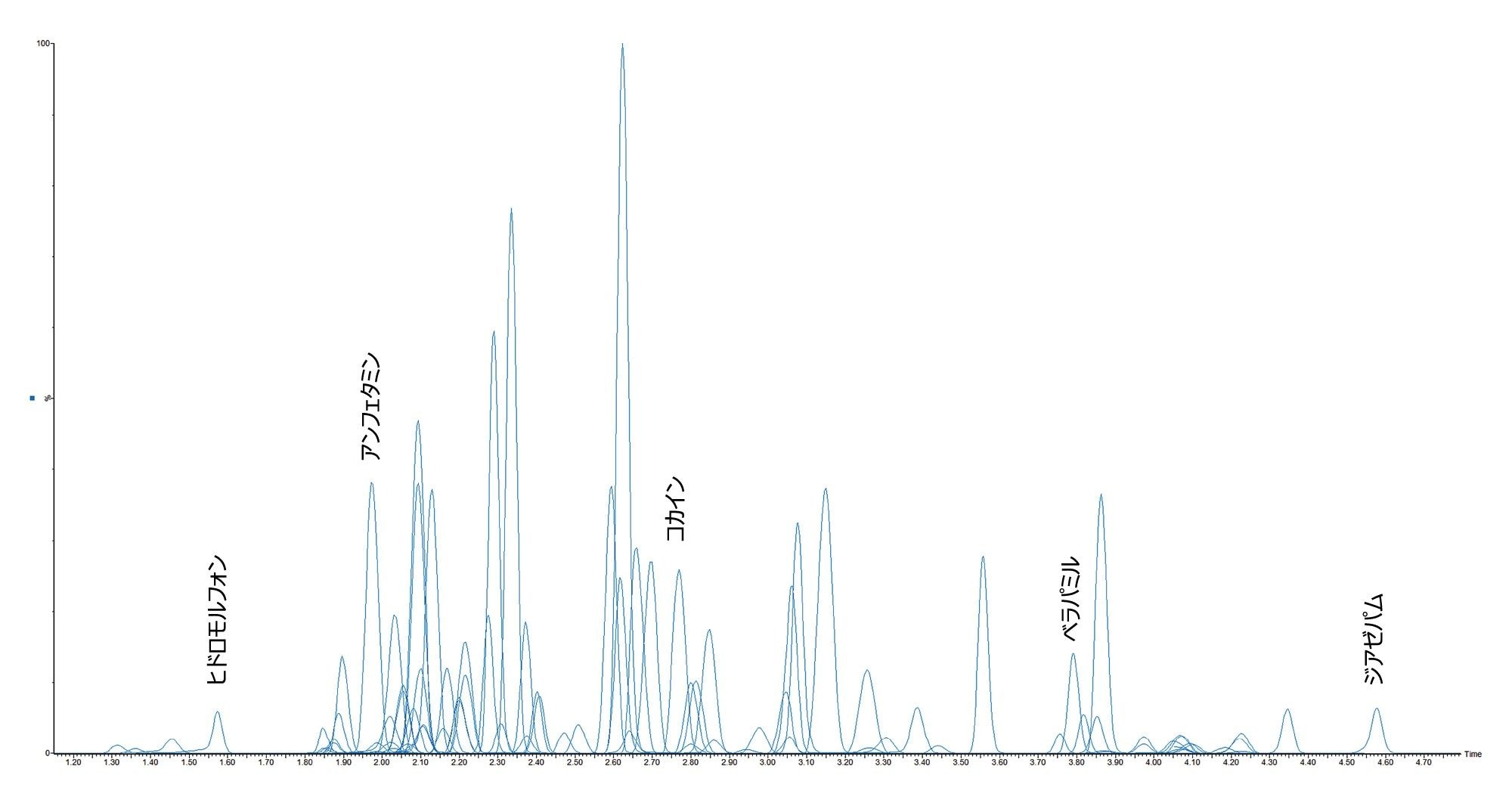
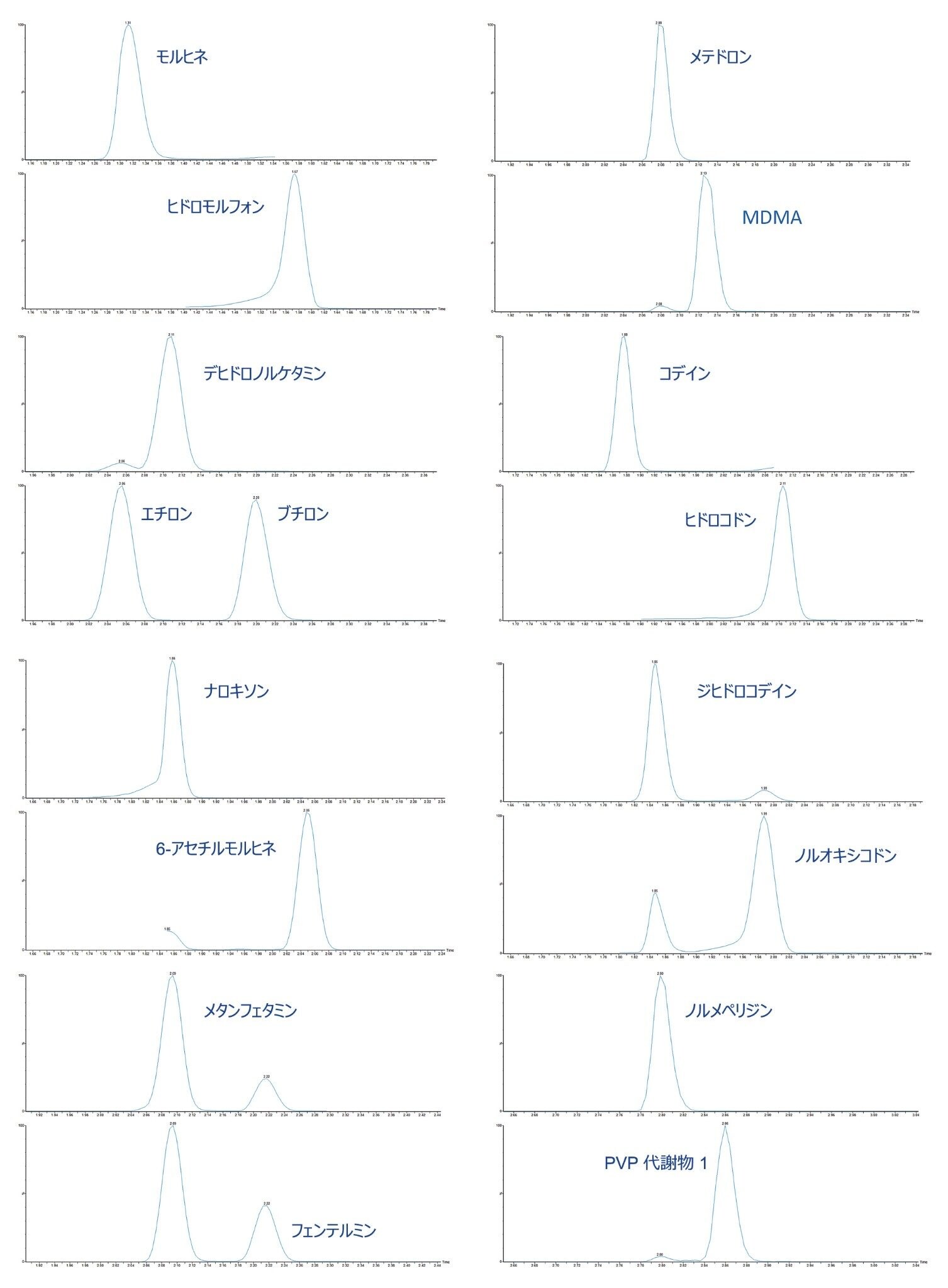
すべての試験化合物および保持時間ウィンドウ、キャリブレーション範囲が付録 1 に記載されています。図 2 に、すべての化合物のクロマトグラフィー分離の重ね描きを示し、図 3 に、相互に干渉する可能性のある分析種の複数のグループからのクロマトグラムを示します。これらは以下に一覧表示しています。
|
モルヒネとヒドロモルフォン |
メテドロンと MDMA |
|
デヒドロノルケタミン、エチロン、ブチロン |
コデインとヒドロコドン |
|
ナロキソンと 6-アセチルモルヒネ |
ジヒドロコデインとノルオキシコドン |
|
メタンフェタミンとフェンテルミン |
ノルメペリジンと PVP 代謝物 1 |
示したケースすべてにおいて、ベースラインクロマトグラフィー分離が得られているため、あるいは選択的 MRM を使用しているために、これらの化合物は相互に干渉しません。この希釈メソッドはサンプルクリーンアップの選択性が低いため、アプリケーションノート 720006187JA と比較すると、UPLC 分離がわずかに変化していました。グラジエントの開始時に一時的にホールドすることで塩およびその他のマトリックス成分を廃液に転流させてから、分析種をカラムから溶出させました。
7 点検量線を 2 回繰り返しで抽出し、各分析実行について分析しました。高濃度範囲のキャリブレーション試薬は 25 ~ 2,500 ng/mL、中濃度範囲のキャリブレーション試薬は 10 ~ 1,000 ng/mL、低濃度範囲のキャリブレーション試薬は 2 ~ 200 ng/mL で、詳細は付録 1 に示しています。すべての検量線で相関係数 (r2)が 0.99 超であり、N-デスメチルゾピクロン、ノルプロポキシフェン、ゾピクロンを除き、少なくとも 75% の検量線ポイントが公称値の ±15%(キャリブレーション試薬 1のレベルでは ±20%)以内でした。キャリブレーション試薬を調製する際に溶解度/安定性を改善できるかどうかを確認するには、これらの化合物についてさらに調査が必要です。N-デスメチルゾピクロン、ノルプロポキシフェン、ゾピクロンについては、定性的なモニタリングのみを行いました。
高濃度範囲の QC サンプルの濃度は 25、75、187.5、1875 ng/mL、中濃度範囲の QC サンプルの濃度は 10、30、75、750 ng/mL、低濃度範囲の QC サンプルの濃度は 2、6、15、150 ng/mL で、詳細は付録 1 に示しています。QC サンプルの少なくとも 66% は、すべての分析において公称値の ±15%(QC1 レベルでは ±20%)以内でした。
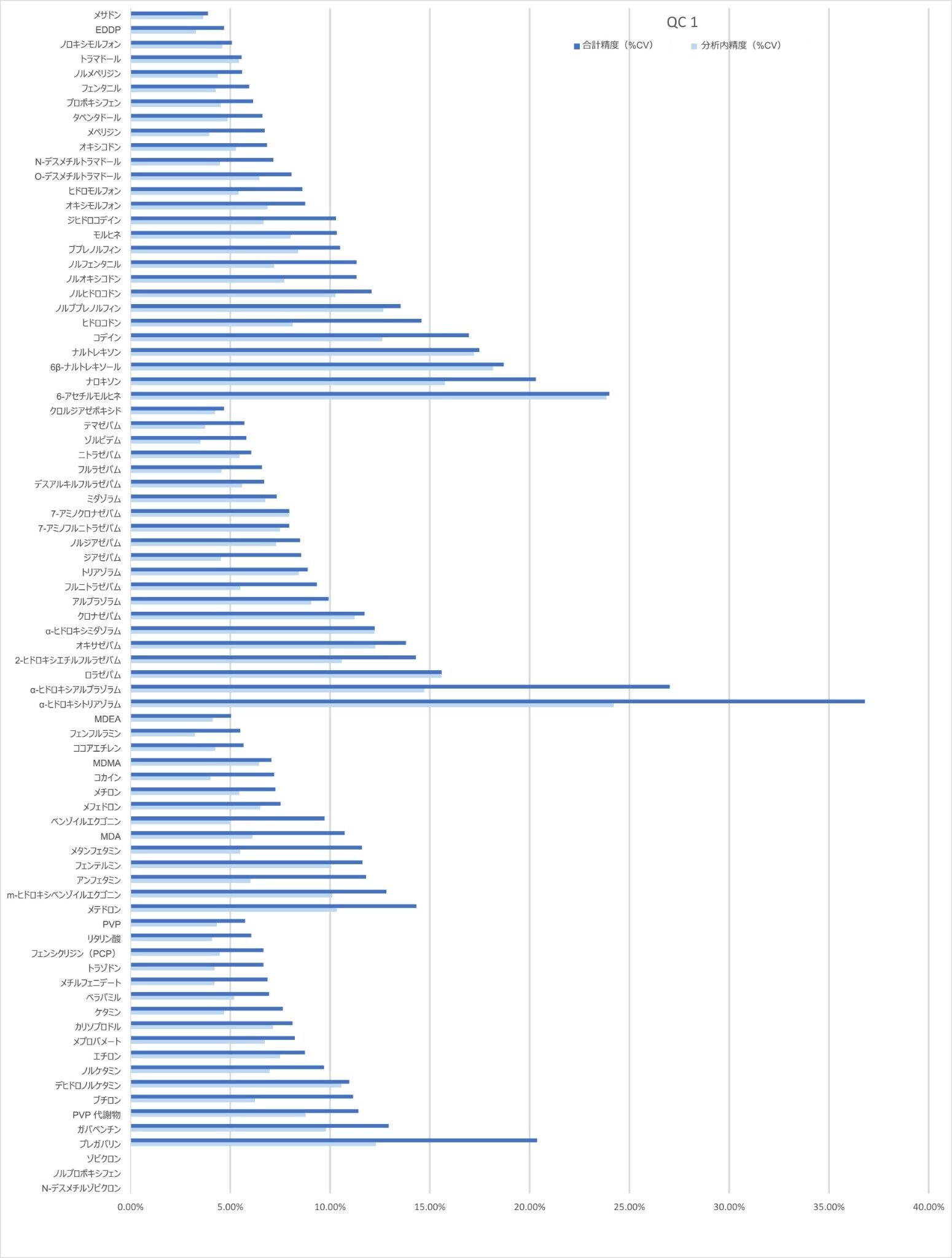
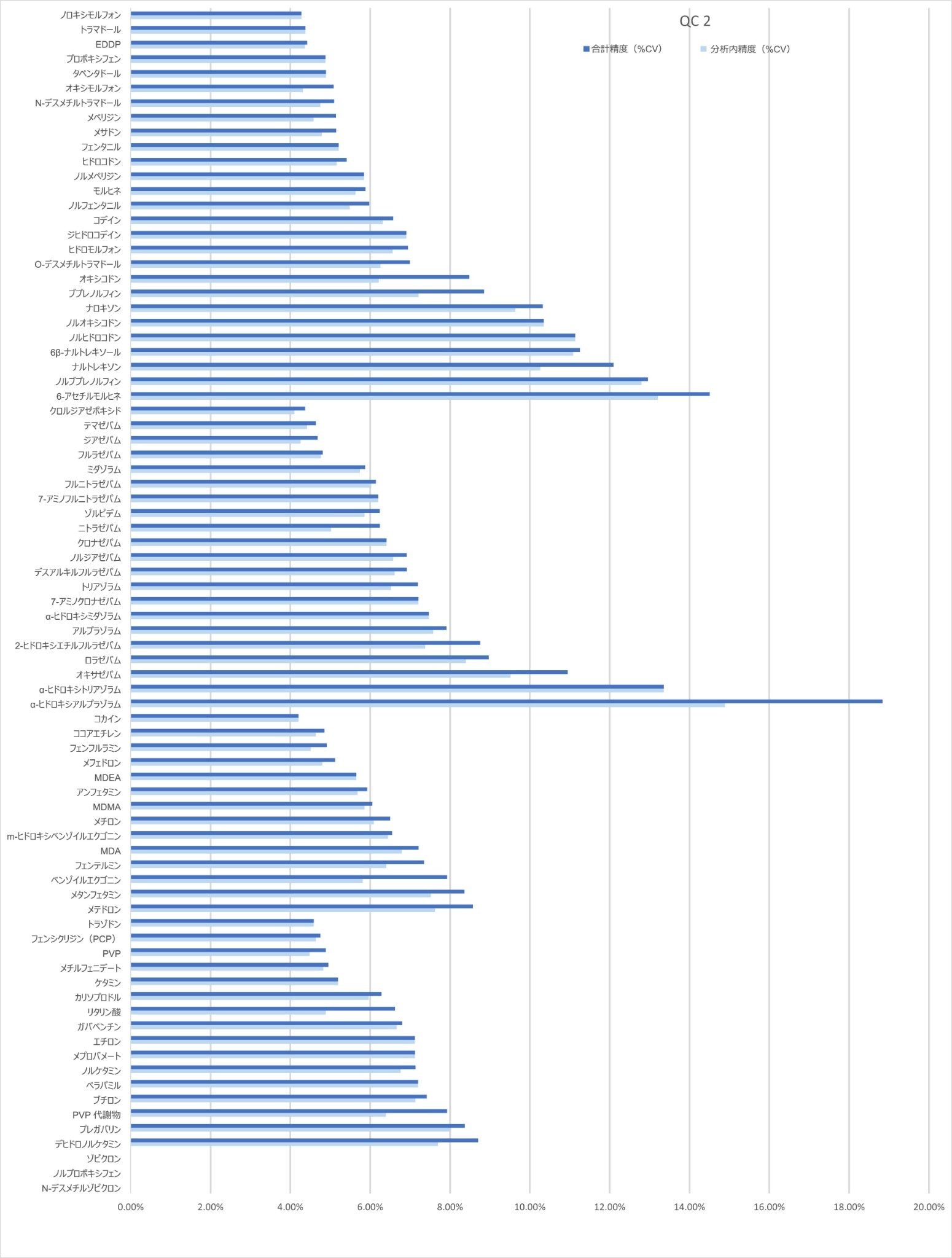
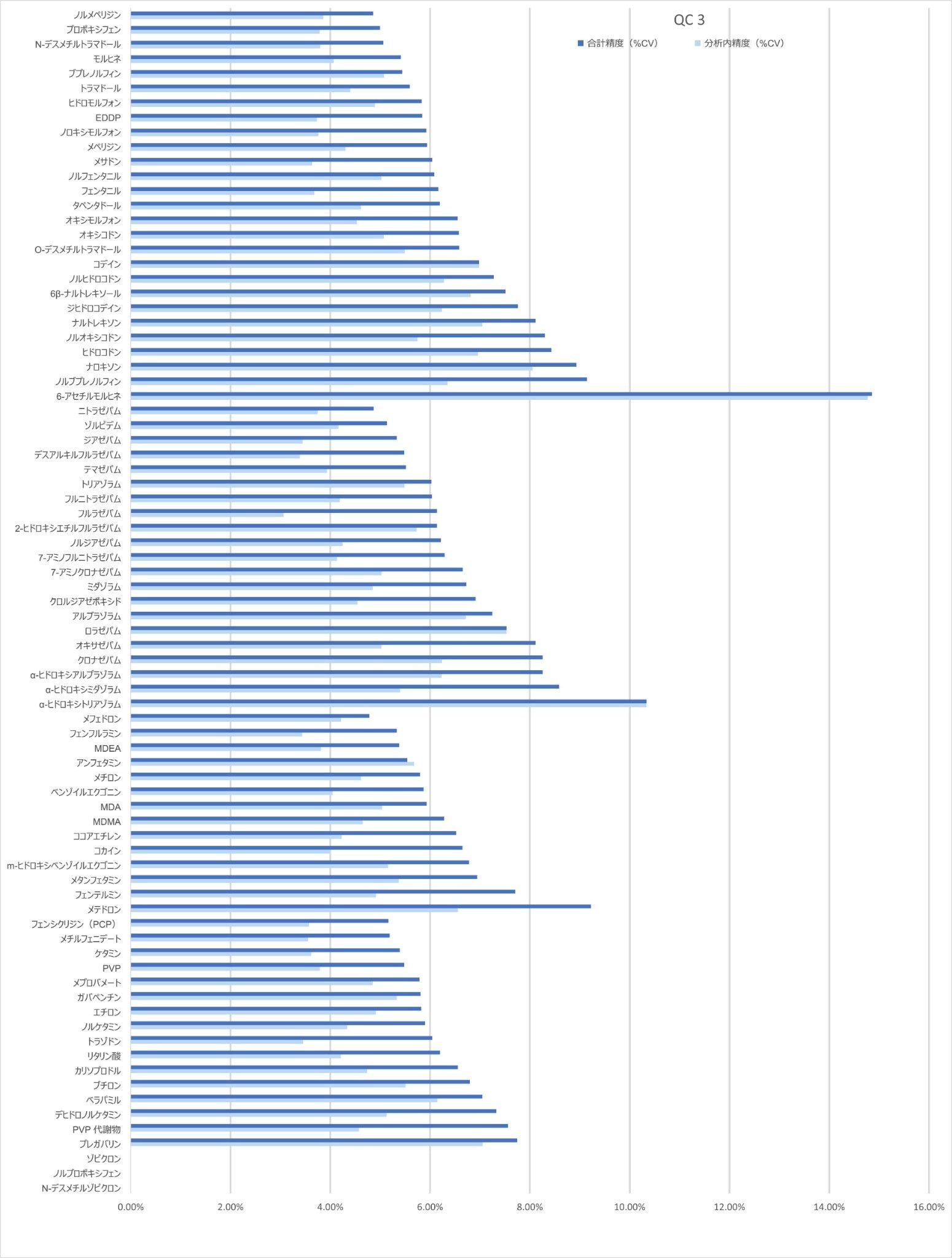
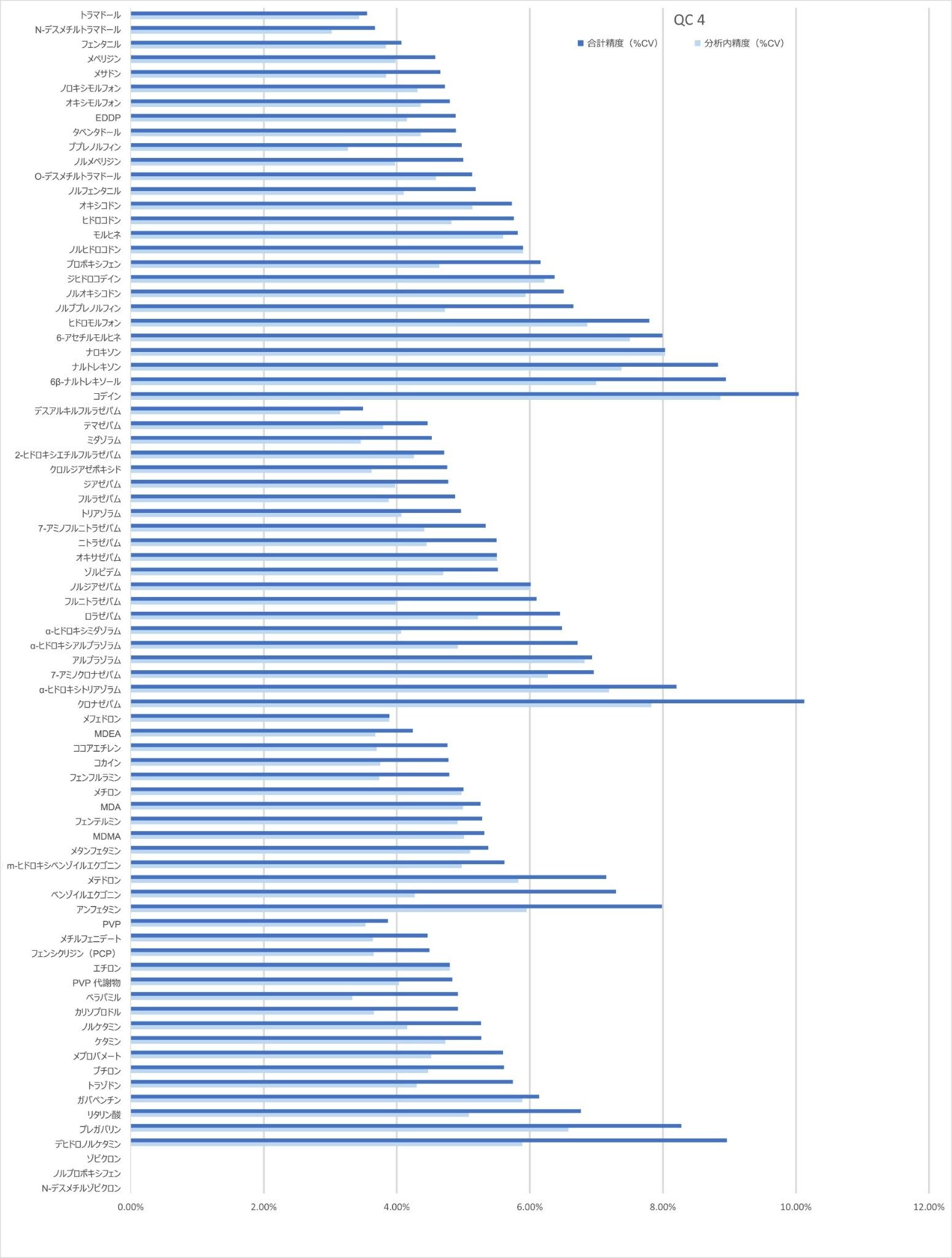
精度性能
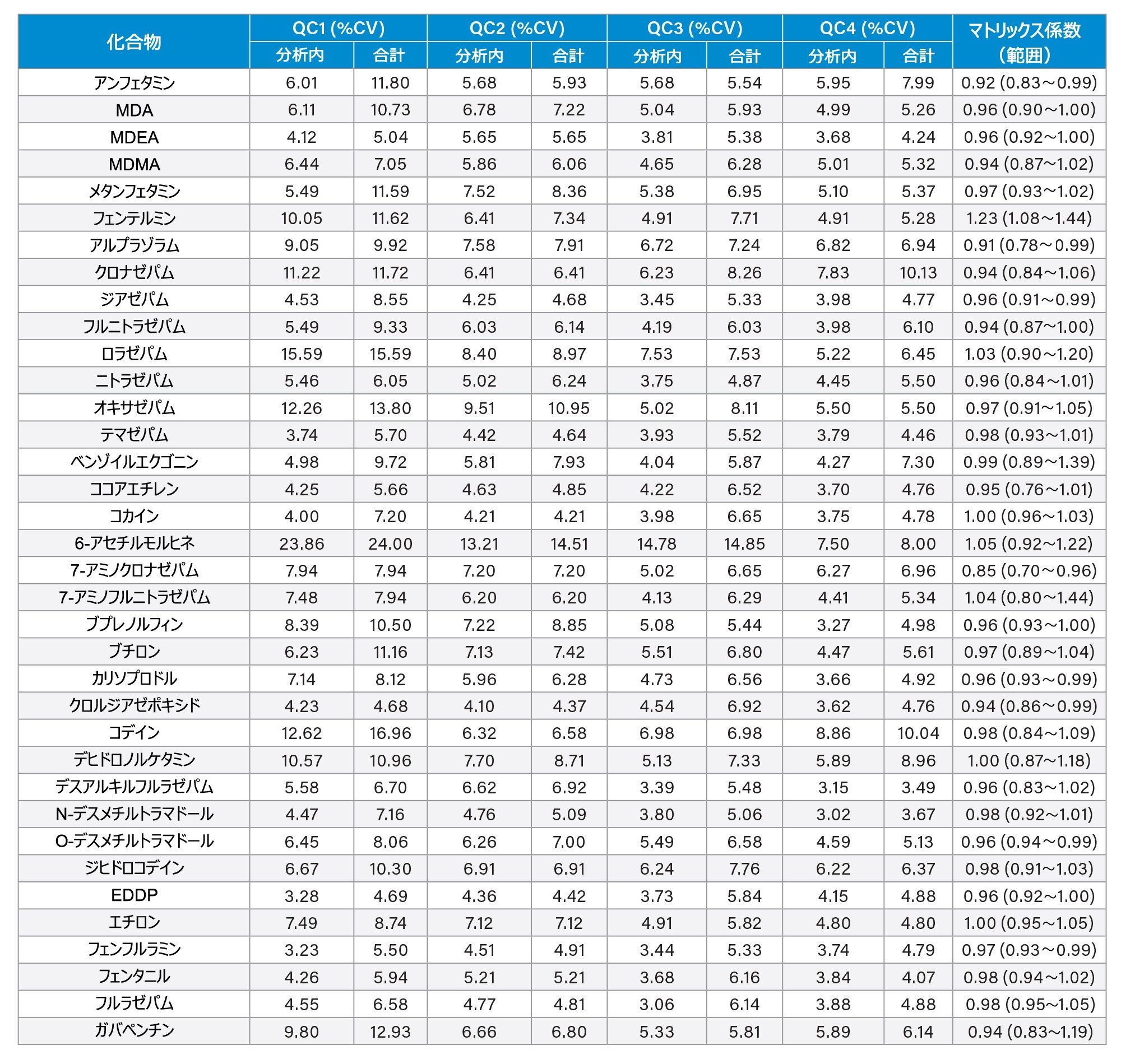
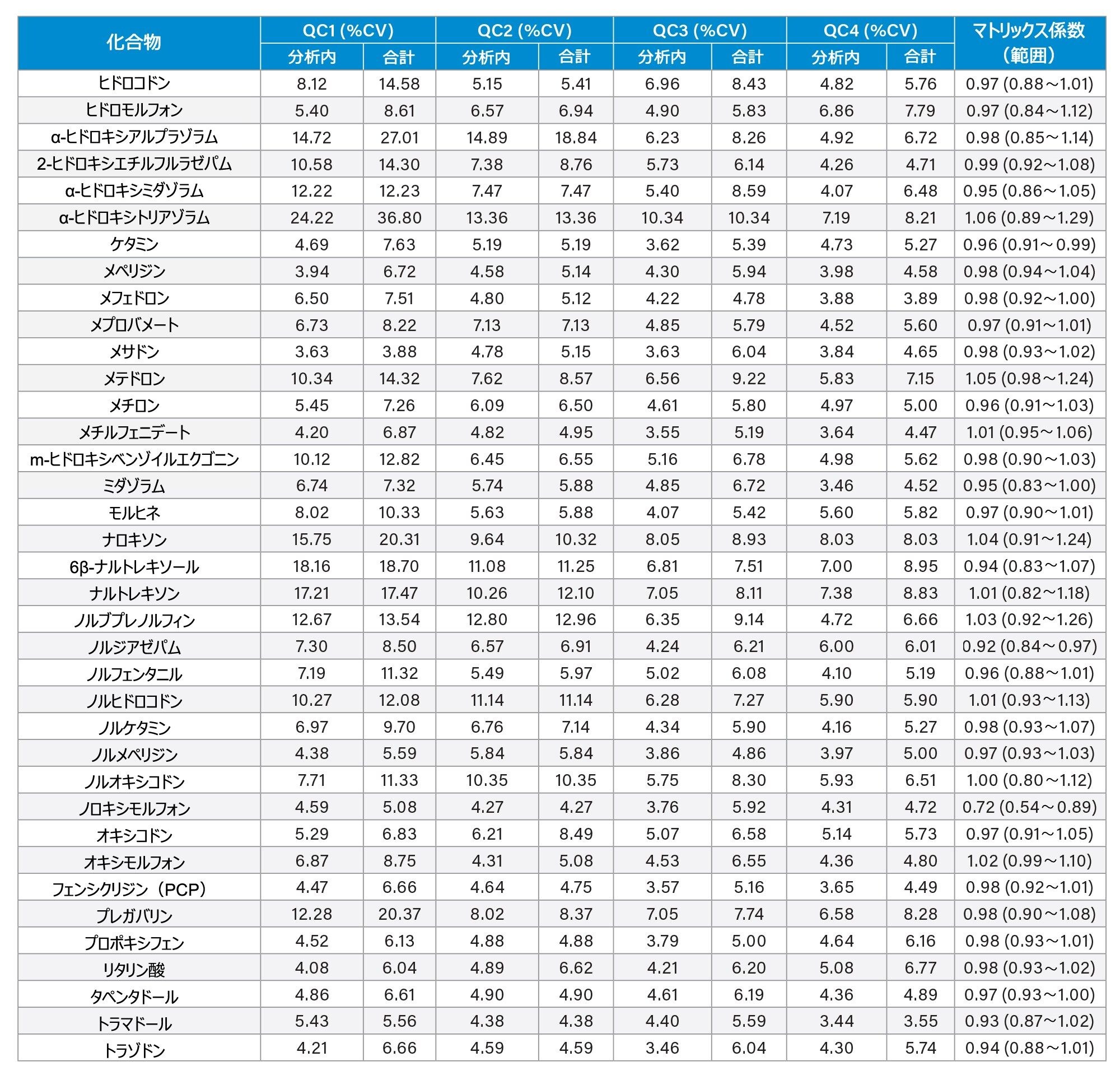
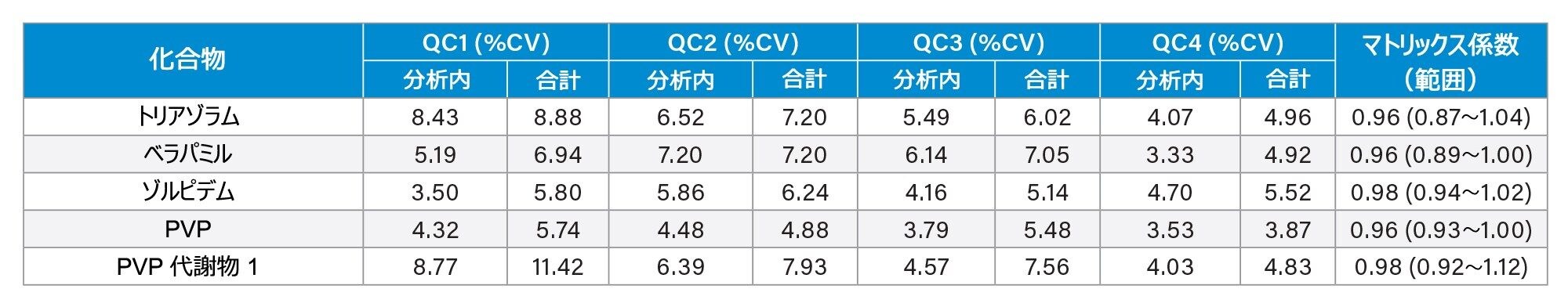
分析法の精度は、各 QC サンプル(濃度は上記)の 5 回繰り返しをそれぞれ 5 回ずつ抽出および分析することにより評価しました(n = 25 の繰り返し)。図 4a ~ 4d に、各 QC レベルで得られた結果のサマリーを示します。
分析感度
キャリブレーション試薬 1 の濃度を下回る低レベルサンプルの 10 回繰り返しをそれぞれ 4 回ずつ抽出および分析して、%CV を計算しました。大部分の分析種について、許容基準 20% CV 未満が得られました。ロラゼパム、6-アセチルモルヒネ、α-ヒドロキシアルプラゾラム、α-ヒドロキシトリアゾラム、ナロキソン、ナルトレキソン、プレガバリンについて、低濃度が必要な場合は、ウォーターズアプリケーションノート 720006187JA に記載しているサンプル抽出メソッドを使用することが推奨されます。
キャリーオーバー
6 つのブランクマトリックスサンプルの平均を、キャリブレーション試薬 7 の 2 倍濃度のサンプルの後に注入した 6 つのブランクマトリックスサンプルの平均と比較したところ、ほぼすべての分析種について、有意なキャリーオーバーは観察されませんでした。ただし、7-アミノクロナゼパムと 7-アミノフルニトラゼパムは例外で、いずれもキャリブレーション試薬 1 のサンプルピーク面積の 38.8% のキャリーオーバーがありました。これらの分析種のキャリーオーバーがほとんどなくなる濃度を評価するには、さらに試験が必要です。
マトリックス係数
6 人の別々の人から抽出した尿サンプルと、コントロールのサンプル水溶液および低濃度および高濃度になるようにスパイクを行ったサンプルを比較したところ、フェンテルミンとノロキシモルフォンを除くすべての分析種について、マトリックス係数は 0.85 ~ 1.15(マトリックス効果 15% 以内)でした。すべての分析種についてのマトリックス係数のデータのサマリーを付録 2 に記載しています。
結論
このアプリケーションブリーフでは、法中毒学における疼痛管理薬および乱用薬物の UPLC-MS/MS 分析について概説しています。サンプル前処理が直接希釈注入アプローチに簡素化され、迅速な抽出手法が実現します。UPLC 分離および Xevo TQ-S micro IVD による検出と組み合わせた、疼痛管理薬および乱用薬物の大規模なパネルに対応する分析法が示されました。結果は精密で、この大規模なパネル内の大部分の分析種について、マトリックス効果は一貫しており、キャリーオーバーは最小限であることがわかりました。特定の化合物については、分析感度を向上させるために、ウォーターズアプリケーションノート 720006187JA に記載した手法のような、よりしっかりしたサンプル抽出手法が推奨されます。
参考文献
- Danaceau JP, Freeto S, Calton L. A Comprehensive Method for the Analysis of Pain Management Drugs and Drugs of Abuse Incorporating Simplified, Rapid Mixed-Mode SPE with UPLC-MS/MS for Forensic Toxicology.Waters Application Note 720006187.2019 March.
- Rosano TG, Rumberger JM, Wood M. Matrix Normalization Techniques for Definitive Urine Drug Testing.Journal of Analytical Toxicology, 2021;45:901–912.
Appendix 1
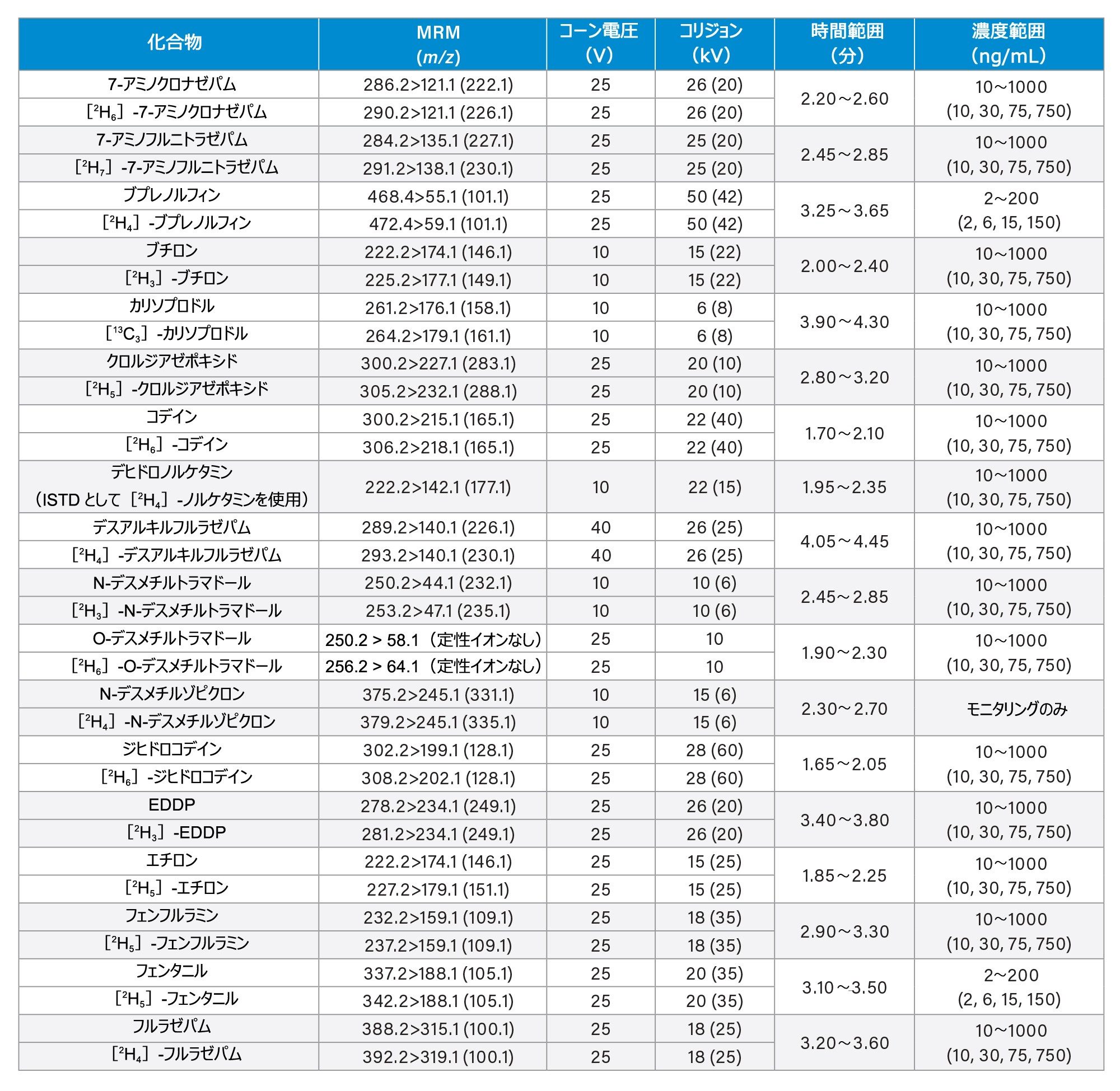
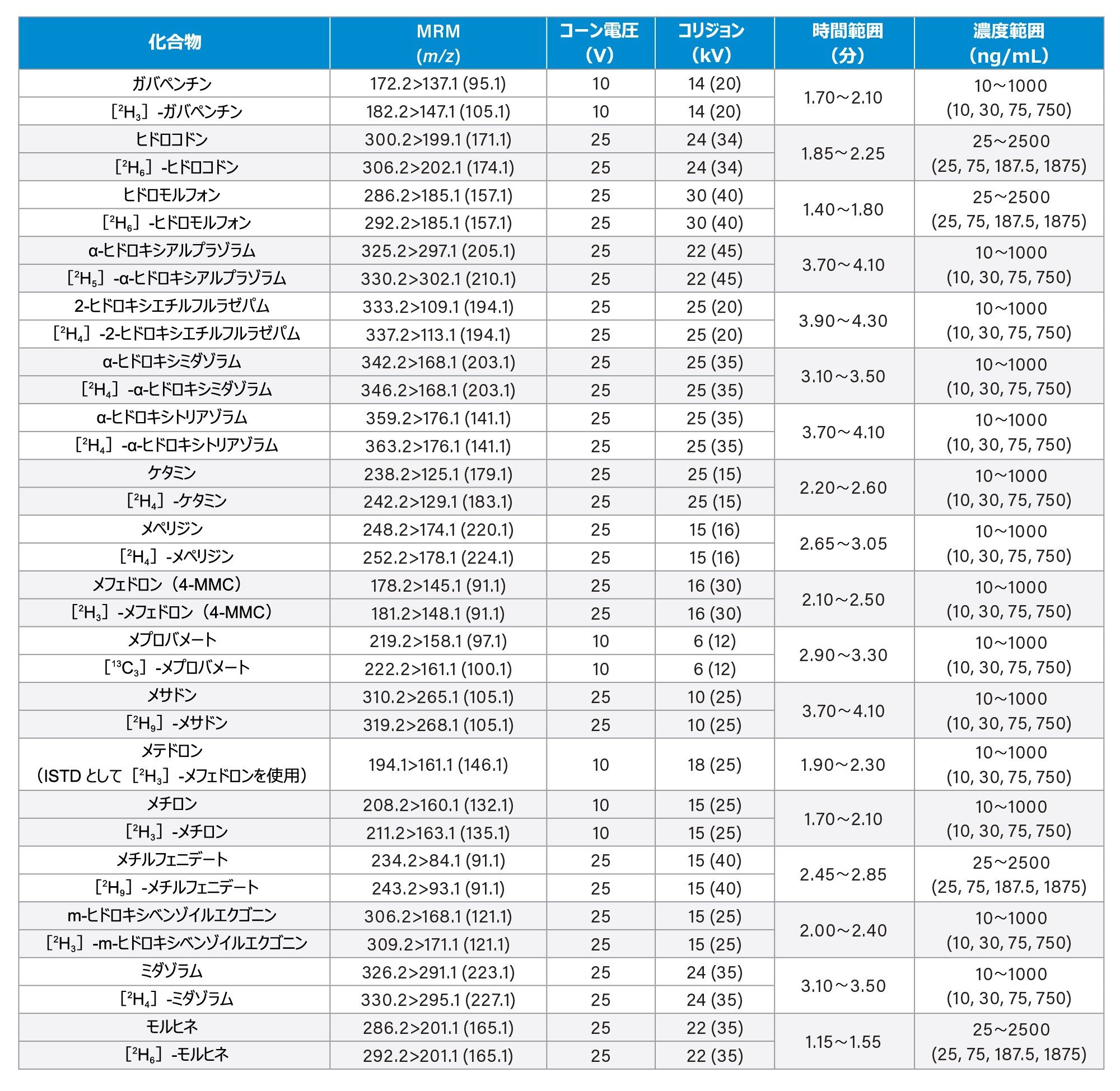
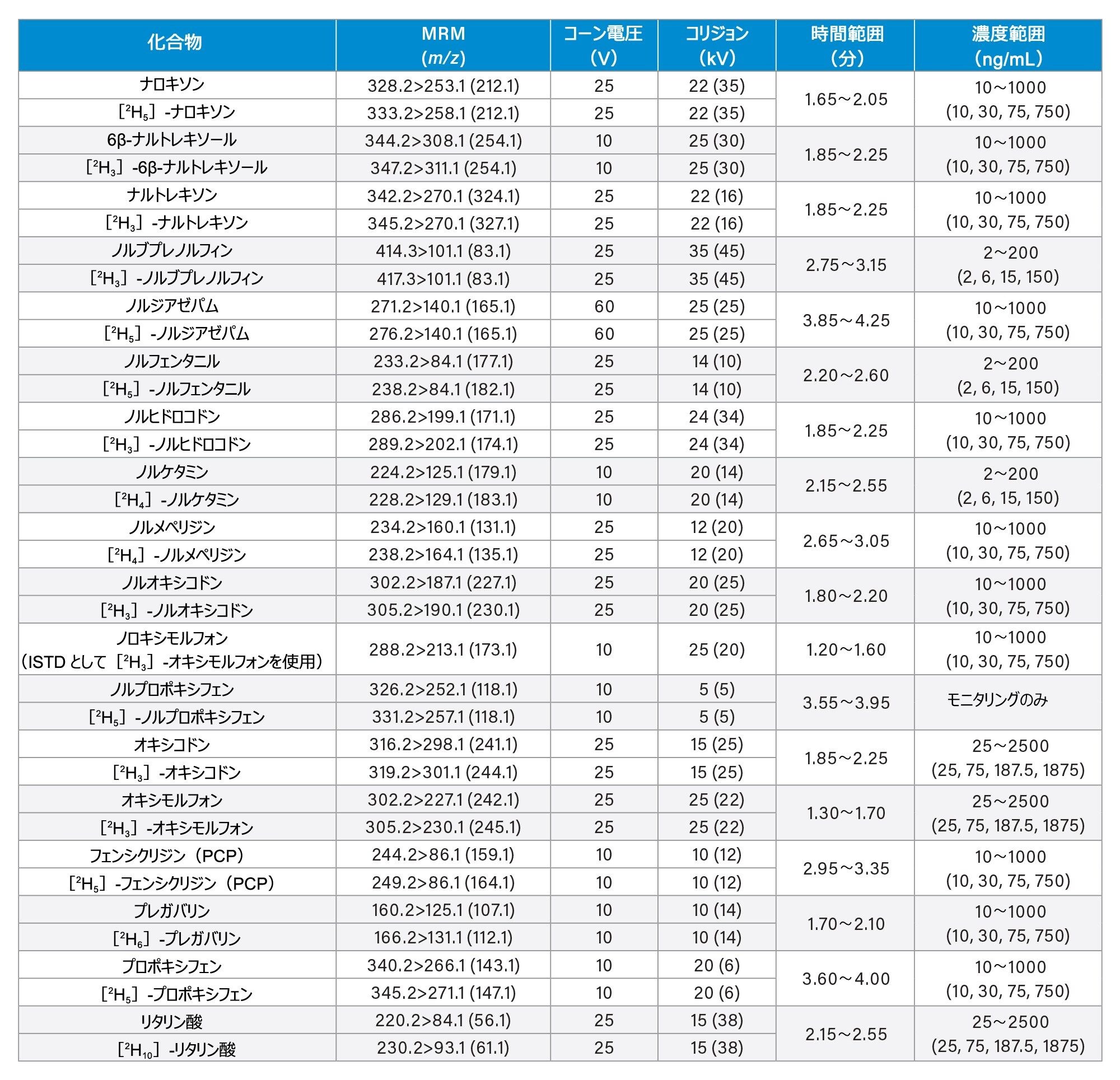
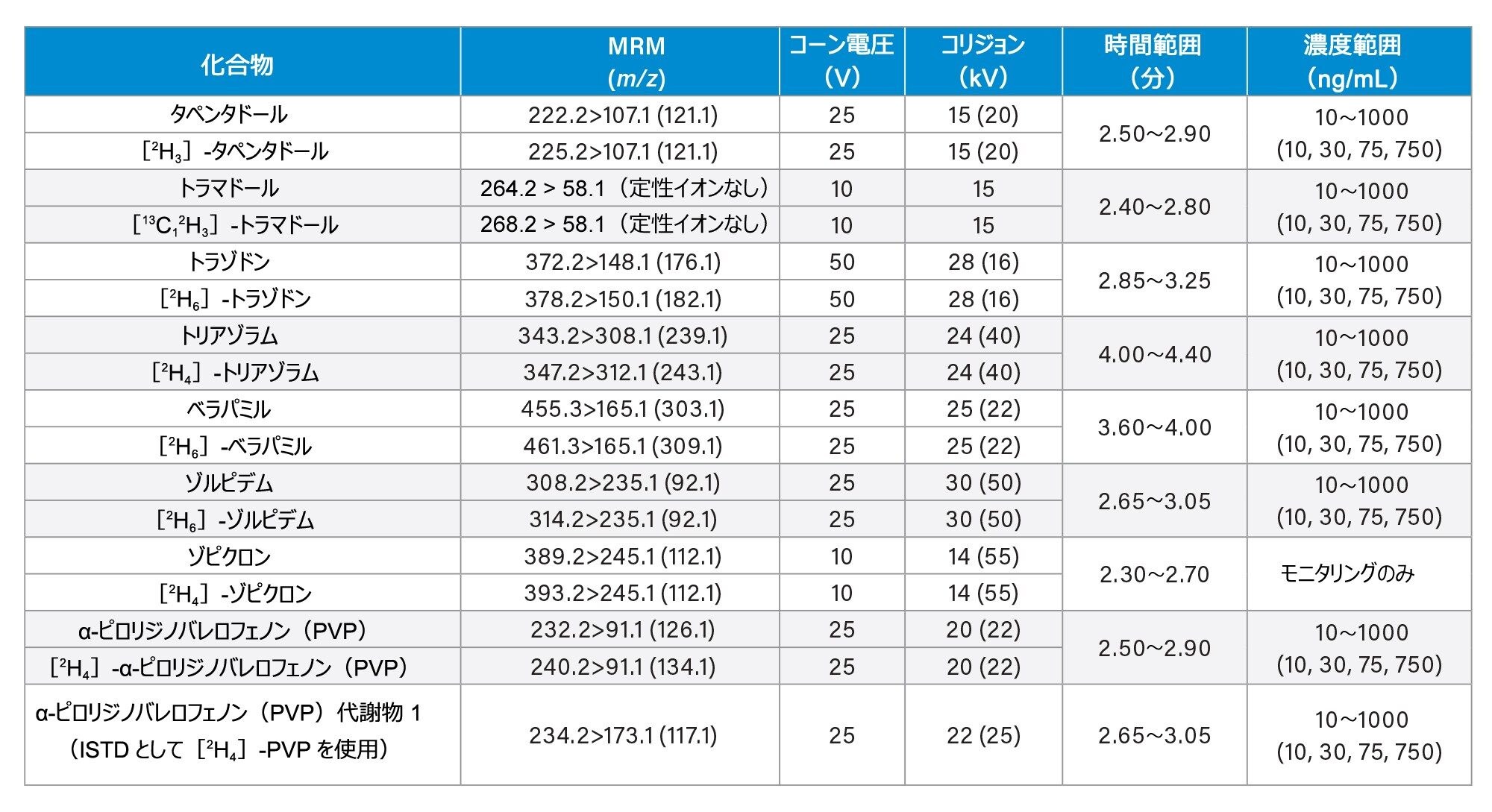
Appendix 2
720007898JA、2023 年 6 月