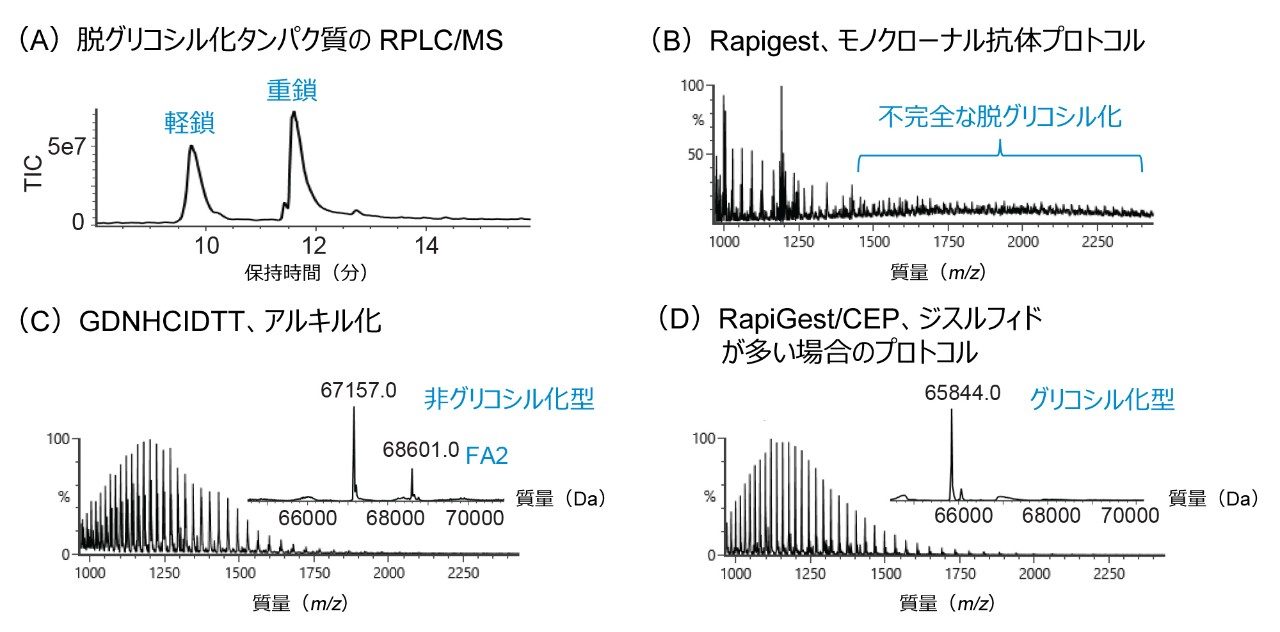
融合タンパク質は、モノクローナル抗体(mAb)と比べて、血漿の半減期延長や治療効果の持続などの利点があるバイオ医薬品の新たなモダリティです1。主要なクラスの融合タンパク質は一般に、構造的に、IgG Fc 領域、および受容体またはリガンドに由来するターゲット結合領域で構成されています2。互いに密接に関連している mAb と比べ、Fc 融合タンパク質には通常、追加のグリコシル化部位があり、ターゲット結合領域の糖鎖構造が複雑であるため、糖鎖プロファイルがより複雑になります。N-グリコシル化レベルは、融合タンパク質医薬品の安全性、有効性、および薬物動態に影響を及ぼす可能性があるため、糖鎖プロファイルの正確な特性解析とモニタリングが、製品の品質と一貫性を保つために非常に重要になります2。タンパク質バイオ医薬品における N-グリコシル化の詳細な情報を得るための有効な方法として、誘導体化および LC-蛍光(FLR)-MS による遊離糖鎖解析が用いられてきました。しかし、融合タンパク質に一般的に見られる重度のジスルフィド結合が、内部に埋もれたグリコシル化部位へのペプチド-N-グリコシダーゼ F(PNGase F)のアクセスを妨げ、その結果、糖鎖の遊離が不完全になる場合があります。このようなジスルフィドリッチ構造は、複雑な糖鎖プロファイルに加え、mAb ベースの遊離糖鎖解析ワークフローを適用する際の課題となっています。そのため、ジスルフィドリッチ融合タンパク質の N-グリコシル化の特性解析とモニタリングを改善するために、遊離糖鎖解析の効率的なワークフローが非常に強く求められています。
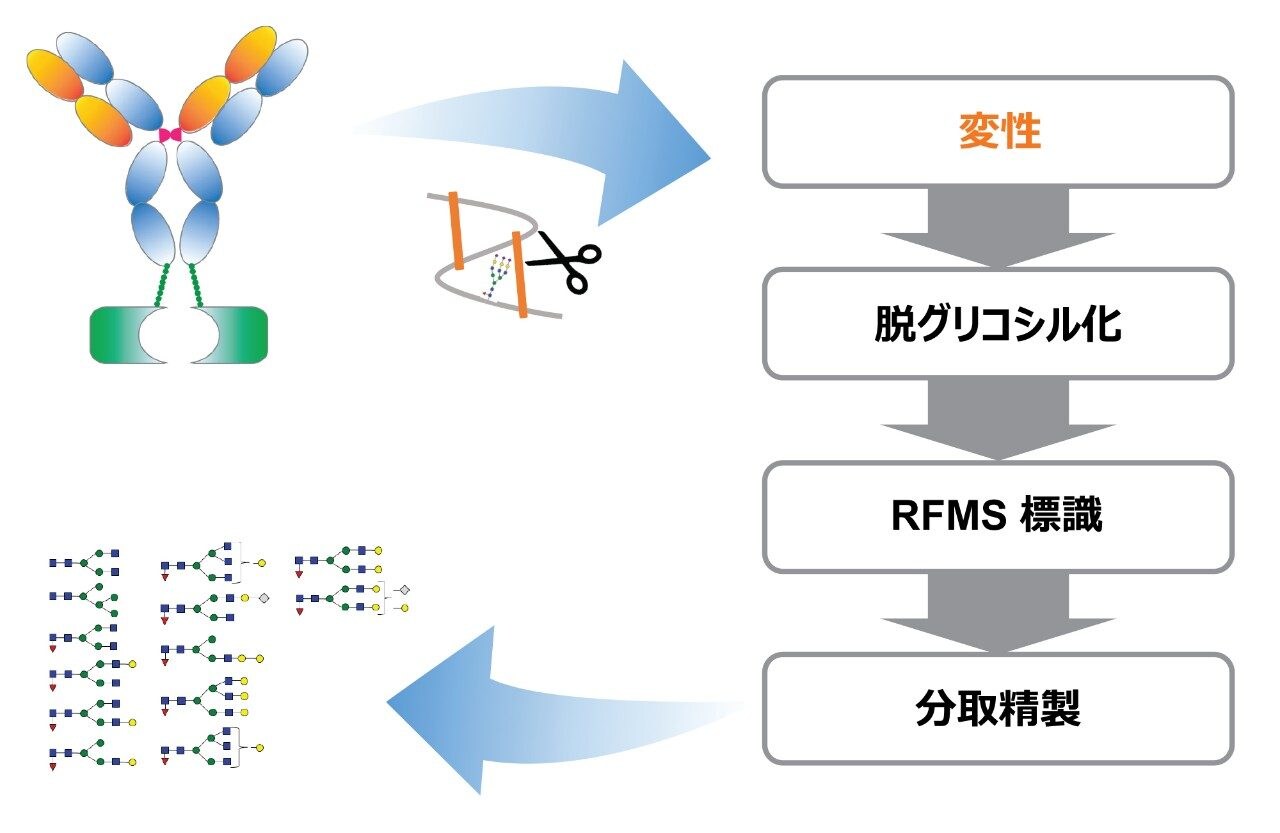
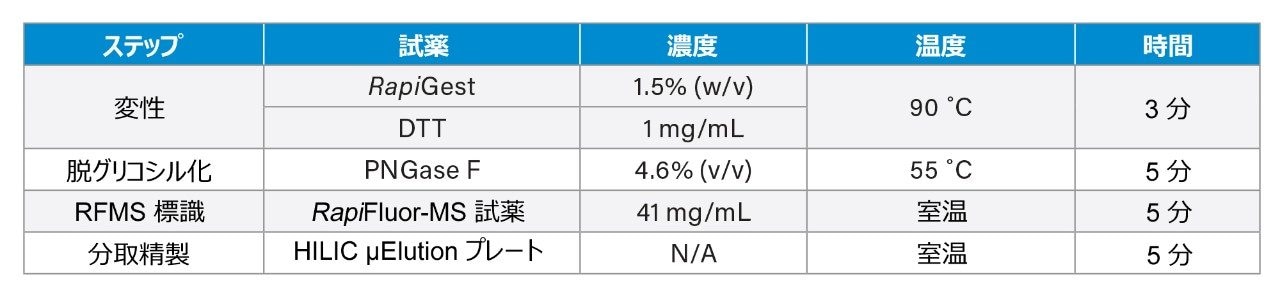
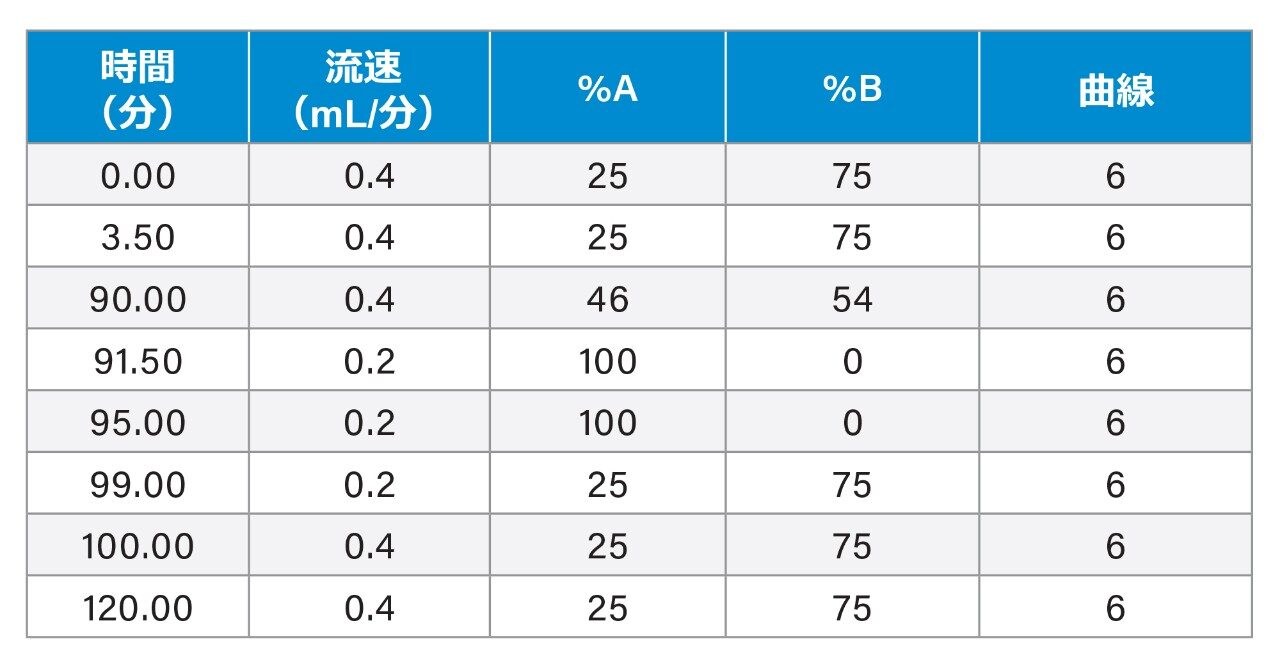
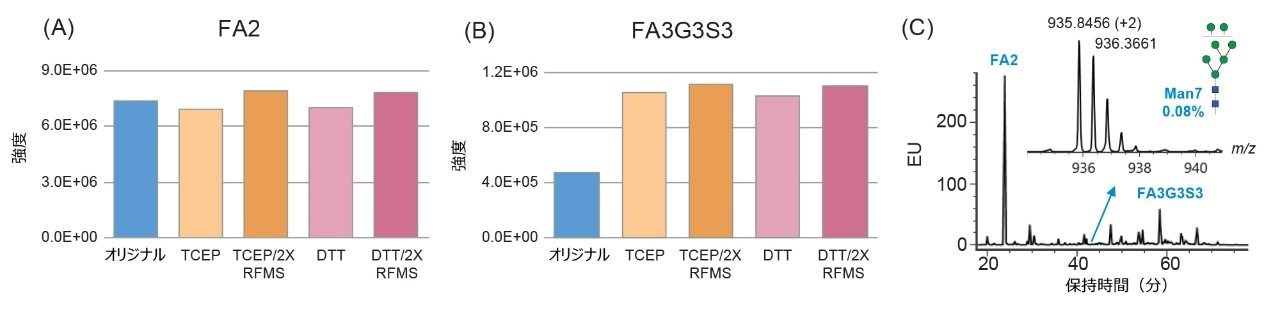
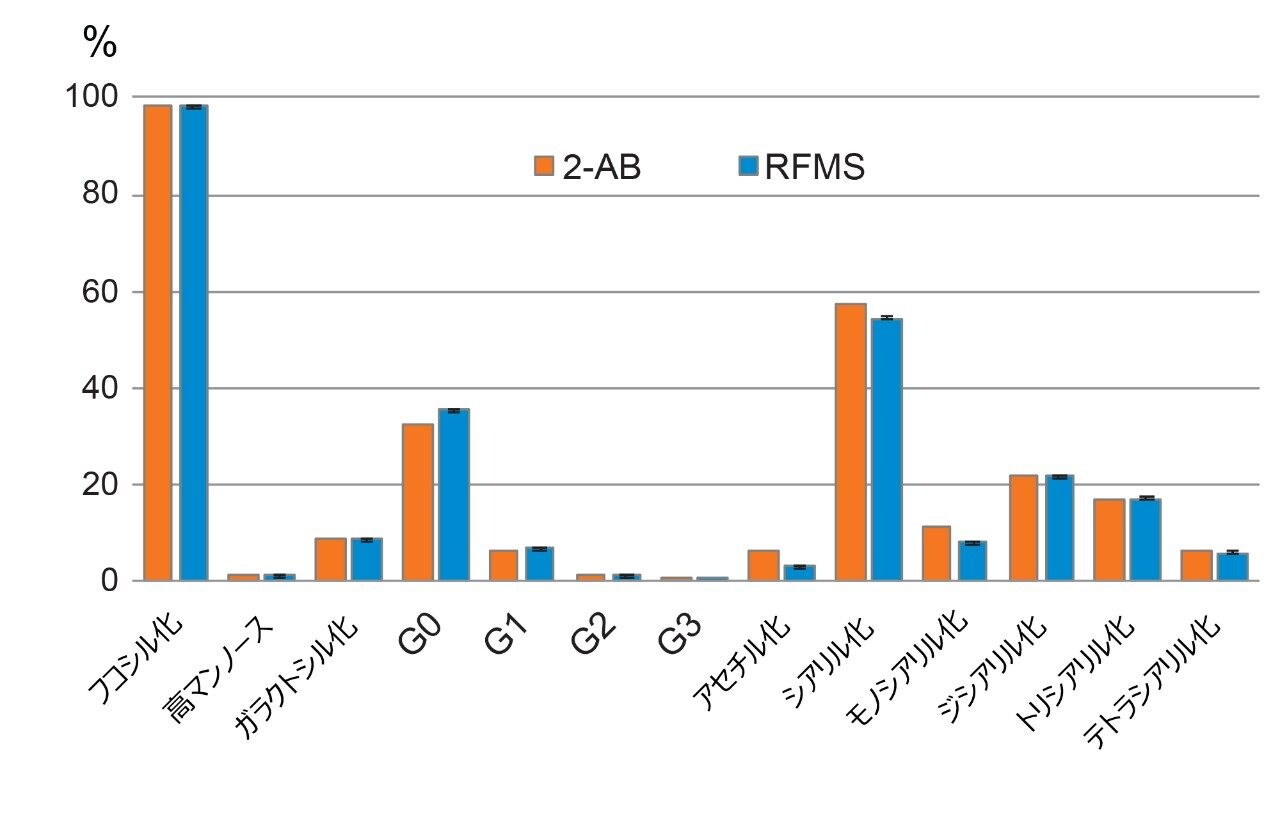
ここでは、Waters GlycoWorks RapiFluor-MS N-Glycan キットのプロトコルを調整し、高レベルのジスルフィド結合と複雑な糖鎖プロファイルを有する融合タンパク質の UPLC-FLR-MS ベースの N 型糖鎖解析を改善するための新しいサンプル前処理法を報告します。迅速な遊離・標識キットとして Waters GlycoWorks RapiFluor-MS N-Glycan キットを選択しました。このキットは、遊離 N 型糖鎖の FLR および MS 検出における大幅なシグナル強度の向上を維持しつつ、最小限のプロトコルの変更で、ジスルフィドリッチ融合タンパク質に容易に適応させることができます3。 調整されたプロトコルの一環として、変性ステップに還元剤を添加してジスルフィド結合を還元し、PNGase F 酵素が N 結合糖鎖にアクセスしやすくなるようにしました(図 1)。この改良したサンプル前処理法を、BioAccord System による効率的な LC-FLR-MS 遊離糖鎖ワークフロー4 と組み合わせることで、ジスルフィドリッチ融合タンパク質の開発および製造プロセス全体にわたって N 型糖鎖解析の精度と効率を向上させることができます。