UltraPerformance コンバージェンスクロマトグラフィー(UPC2)によるコレステロール低下薬の分析における多製品対応分析法の開発
要約
この文書では、ACQUITY UltraPerformance コンバージェンスクロマトグラフィー(UPC²)システムを使用した、実験計画法アプローチに基づく分析法開発の各ステップを説明しています。試験した化合物は、血清中の高コレステロール値を下げるのに使用されるスタチン系に属しています。複数の医薬品処方においてスタチン系と組み合わせて使用されているエゼチミブを追加しました。まず、5 種のカラムを様々なモディファイヤーおよびサンプル希釈液で評価しました。ACQUITY UPC² カラム設計を採用した Torus 1-アミノアントラセン(1-AA)カラムは、すべての分析種の分離において最高の性能を示しました。次に、中心複合計画(Central Composite Design、CCD)を用いて、圧力、温度、モディファイヤーの量が微調整パラメーターとして分離に及ぼす影響を評価しました。最終的なクロマトグラフィー条件として、Torus 1-AA カラム、モディファイヤーとしてエタノール:水(95:5)、希釈液としてアセトニトリル-エタノール(3:2)、流速 1.80 mL/分、オーブン温度 42 ℃、背圧レギュレーター 2175 psi、4 分で 5% ~ 15.5% モディファイヤーのグラジエントを使用して測定した結果、クリティカルペアの最小分離度 2.0 という最良の分離が得られました。ブラジルの規制機関(ANVISA)の指示に従って分析法のバリデーションを行ったところ、r > 0.99、正確性の範囲 95% ~ 105%、精度 4.4% 以下(日内および日間)になり、分析種の濃度に照らして許容範囲内でした。
アプリケーションのメリット
- コレステロール低下薬の分析のための迅速な ACQUITY UPC2 分析法
- 環境に優しい溶媒であるエタノールを少量使用し、移動相に添加剤が不要な、酸性コレステロール低下薬の分析
- 主要な移動相として二酸化炭素を使用することで、有機溶媒の消費量を削減
はじめに
世界保健機関(WHO)が 2016 年に実施したパネルでは、世界の主な死因として、主に高所得国において、第 1 位に虚血性心疾患、第 2 位に脳卒中が挙げられています1。 これらの疾患は、血清中のコレステロールの増加に関連しています。スタチン系は、肝細胞でのコレステロール合成に関わるヒドロキシメチルグルタリルコエンザイム A(HMG-CoA)還元酵素の作用を阻害することで、コレステロール値を低下させることが知られています2。 そのため、スタチン系は高コレステロール血症の治療に幅広く使用されています。治療法によっては HMG-CoA の作用を阻害することでコレステロール値が適切に低下しない場合があるため、小腸でのコレステロールの吸収を減少させる薬剤であるエゼチミブが、スタチンと組み合わせて使用され、効果を挙げています3。
通常は、各スタチンあるいはエゼチミブとの組み合わせに対して分析法が開発されています4–7。 これらの医薬品のほとんどに対して逆相液体クロマトグラフィー(RP-LC)が使用されており、有機溶媒(アセトニトリル、メタノール)が大量に消費され、酸性添加剤が使用されています。この文書では、粒子径 2 μm 以下の充塡剤を用いたカラムを使用した超臨界流体クロマトグラフィー(SFC)による迅速な多製品対応の分析法8 により、溶媒の消費量が削減され、添加剤が不要になることを説明しています9。多製品対応分析法という用語は、同じ製剤に含まれるかどうかに関わらず、複数の薬剤に使用できる単一の分析法開発を指します。
SFC では超臨界流体(通常は二酸化炭素、CO2)を移動相として使用します。超臨界 CO2 を移動相とする利点として、低粘度で溶媒和能が高く、気相と液相の中間であり、環境に優しいと言われている点が挙げられます10。 加圧 CO2 の供給の改善と、自動背圧レギュレーター(ABPR)による頑健な圧力制御により超臨界流体を安定化させたことが、超臨界流体クロマトグラフィーを競争力の高い分析手法として復活させる上で画期的な進歩となりました。CO2 に有機溶媒(モディファイヤー)を添加すると、固有の非極性の特性が変化し、極性の高い化合物の溶出が可能になります11。SFC 設計カラムのリリースに伴うこの点に加え、カラム外容量の減少および粒子径 2 μm 以下の使用により、より高速かつ高感度の分析が可能になり、超高速液体クロマトグラフィーと同等以上の分離効率が得られました8。
最新の分析法開発においては、特に分析法のクオリティ・バイ・デザインの枠組みの中で、DOE(実験計画法)ツール12 を用いた実験の変動要因の関数としてのクロマトグラフィーレスポンスのモデル化が強く推奨されています13。この試験では、パラメーターである固定相、移動相モディファイヤーおよびサンプル希釈液をまずマルチレベルカテゴリー計画(Multilevel Categorical Design)で評価し、結果を分析種の化学構造と関連付けました(パート I)。これらは SFC の選択性、ピーク形状、有効性に影響を及ぼす主要変動要因です14。次に、多製品対応分析法ですべての化合物が分離されるようにするため、中心複合計画(Central Composite Design、CCD)を用いて、溶出の強度に影響を及ぼす温度、モディファイヤーの圧力および比率といったパラメーターを試験し、分析法を最適化しました15(パート II)。
実験方法
パート I:
|
システム: |
ACQUITY UPC2 システム |
|
検出: |
PDA 検出器(240 nm で検出) |
|
カラム: |
ACQUITY UPC2: 1)エチレン架橋型ハイブリッド(BEH)、 2)ハイストレングスシリカオクタデシル基結合(HSS C18)、 3)表面チャージハイブリッドペンタフルオロフェニル(CSH PFP)、 4)Torus 1-アミノアントラセン(1-AA)、 5)Torus 2-ピコリルアミン(2-PIC) |
|
移動相 A: |
CO2(ボンベ、医療用グレード) |
|
移動相 B: |
エタノール:H2O 95:5(0 ~ 30 %(v/v)) |
|
カラム温度: |
40 ℃ |
|
ABPR: |
1500 psi |
|
SM 温度: |
室温 |
|
サンプル希釈液: |
アセトニトリル:エタノール 3:2(v/v) |
|
注入量: |
1 μL |
|
流速: |
1.50 mL/分 |
|
バイアル: |
Waters アンバーガラス 12 × 32 mm スクリューネックバイアル、2 mL |
|
PDA スキャン範囲: |
210 ~ 400 nm |
|
強ニードル洗浄溶媒: |
メタノール |
|
弱ニードル洗浄溶媒: |
2-プロパノール:メタノール 1:1 |
|
シール洗浄溶媒: |
メタノール |
パート II:
|
システム: |
ACQUITY UPC2 システム |
|
検出: |
PDA 検出器(275 nm で検出) |
|
カラム: |
ACQUITY UPC2 Torus 1-AA 2.1 × 50 mm、1.7 µm(製品番号:186007623) |
|
移動相 A: |
CO2(ボンベ、医療用グレード) |
|
移動相 B: |
エタノール:H2O 95:5(v/v)、グラジエントの終了は中心複合計画(Central Composite Design)により異なる(CO2 中で 15.5 ~ 27.5%) |
|
カラム温度: |
中心複合計画(Central Composite Design)により異なる(25 ~ 50 ℃) |
|
ABPR: |
中心複合計画(Central Composite Design)により異なる(10.5 ~ 16.3 MPa) |
|
SM 温度: |
室温 |
|
サンプル希釈液: |
アセトニトリル:エタノール 3:2(v/v) |
|
注入量: |
1 μL |
|
流速: |
1.80 mL/分 |
|
バイアル: |
Waters アンバーガラス 12 × 32 mm スクリューネックバイアル、2 mL |
|
PDA スキャン範囲: |
210 ~ 400 nm |
|
強ニードル洗浄溶媒: |
メタノール |
|
弱ニードル洗浄溶媒: |
2-プロパノール:メタノール 1:1 |
|
シール洗浄溶媒: |
メタノール |
妥当な時間で分離が行えるように、最大分析時間を 10 分に設定しました。
サンプルの説明
シンバスタチン、ロバスタチン、ロスバスタチンカルシウム、フルバスタチンナトリウム、アトルバスタチンカルシウム、エゼチミブは、二次医薬品標準試料として Sigma-Aldrich(米国ミズーリ州、EUA)から購入しました。プラバスタチンナトリウムおよびピタバスタチンカルシウムは、Cayman Chemicals(米国ミシガン州、EUA)から純度 98% 以上のものを購入しました。すべての実験において、標準品の混合物をアセトニトリル:エタノール 3:2(v/v)に溶解し(濃度 0.5 mg/mL)、0.22 µm シリンジフィルターでろ過してから注入しました。ブラジル市場の市販品は、最寄りのドラッグストアで調達しました。
結果および考察
パート I:固定相と有機モディファイヤーの選択
分析法開発の最初のステップは、適切な固定相の選択でした。シンバスタチン、ロバスタチン、エゼチミブ以外の分析種は陰イオン性と考えられます。これらの分析種の pKa 値は 5 未満であるため、アルコールモディファイヤーを含む CO2 ベースの移動相の見かけ上の pH が 5 ~ 6 になると言われています16。
図 1 に示すように、ACQUITY UPC2 Torus 1-AA および Torus 2-PIC カラムは、添加剤を必要とせず、陰イオン性および中性の分子種であるシンバスタチン、ロバスタチン、エゼチミブについて高性能を発揮しました。他のカラムでは、陰イオン性分析種が適切なピーク形状で溶出しませんでした。これは、シラノール基に対する反発が原因と考えられます。これらの結果は、SFC 用の固定相の分類のための試験結果と一致していました16,17。さらに、Torus 1-AA のみがシンバスタチン(1)とロバスタチン(2)をわずかに分離することができました18。分子の相違がメチル基 1 つのみであるため、完全に分離することはできないと考えられます。
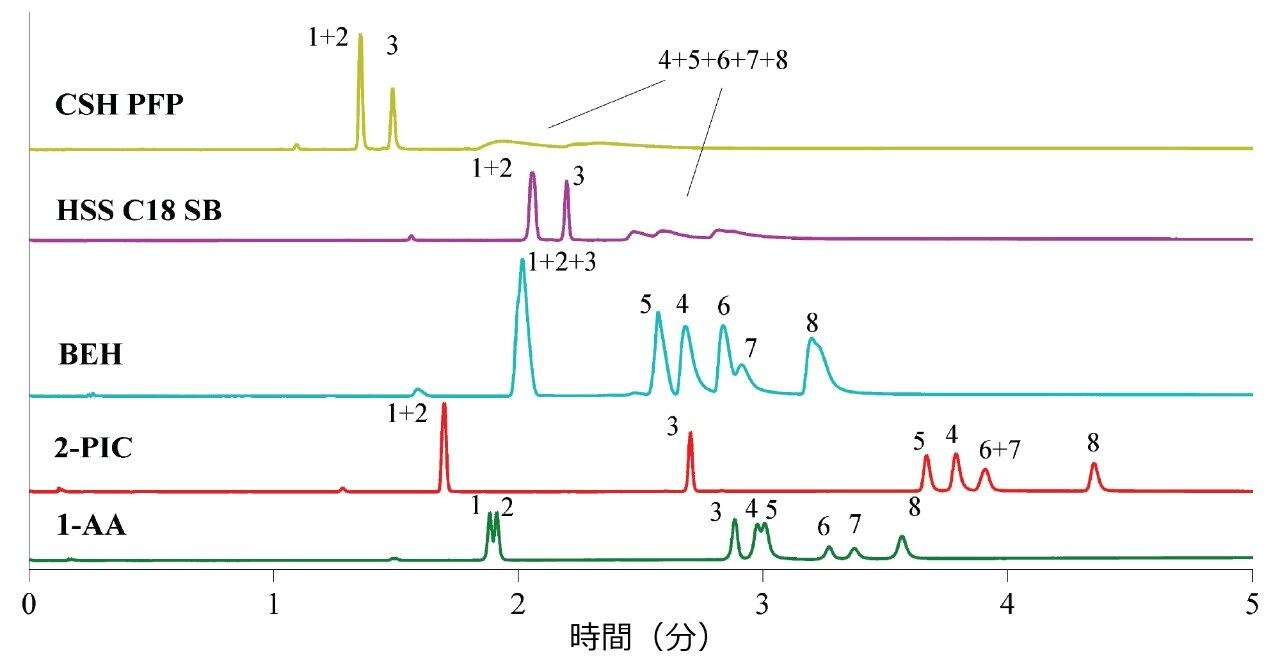
最初のスクリーニングは、5 分間で 0% ~ 30% 有機モディファイヤーの範囲のグラジエント溶出で行いました。メタノール、エタノール、イソプロパノールなどのアルコールの使用による選択性の変化はわずかでしたが、モディファイヤーとしてアセトニトリルを使用した場合、分析時間が長くなり、最大濃度 30% まで超臨界 CO2 の非混和性が見られたことから、アセトニトリルは適していませんでした。これ以降の分析法開発においては、毒性の低さのため、エタノールを選択しました。その後、5 分間で 0 ~ 30% のグラジエント溶出と同じグラジエント勾配を維持するため、グラジエント溶出の初期組成を 5% に変更し、最終組成 25% に 4 分で達するようにしました。グラジエント溶出開始時の濃度が高いほど、移動相の吸着が減少し、溶出強度が高まります。
注入量は 1.0 µL のままにし、希釈液アセトニトリル:エタノール 3:2(v/v)中に溶液を調製しました。超臨界液体クロマトグラフィーにおいて、固定相への溶媒の相互作用を最小限に抑えるために、希釈液としては弱溶媒が望まれます。メタノールなどの強溶媒ではなく、弱溶媒であるアセトニトリルおよびエタノールを使用したのはそのためです。更に、アセトニトリル/エタノール混合液は、これらの分析種にとってより安定した溶液であり、これらを同時に溶解することが可能でした。
このステップの結果から、Torus 1-AA カラム、開始グラジエント組成 5% エタノールモディファイヤー:水 95:5、注入量 1.0 µL、希釈液としてアセトニトリル:エタノール 3:2(v/v)、というクロマトグラフィー条件の使用が適切となりました。
パート II:DoE による超臨界流体の密度の試験
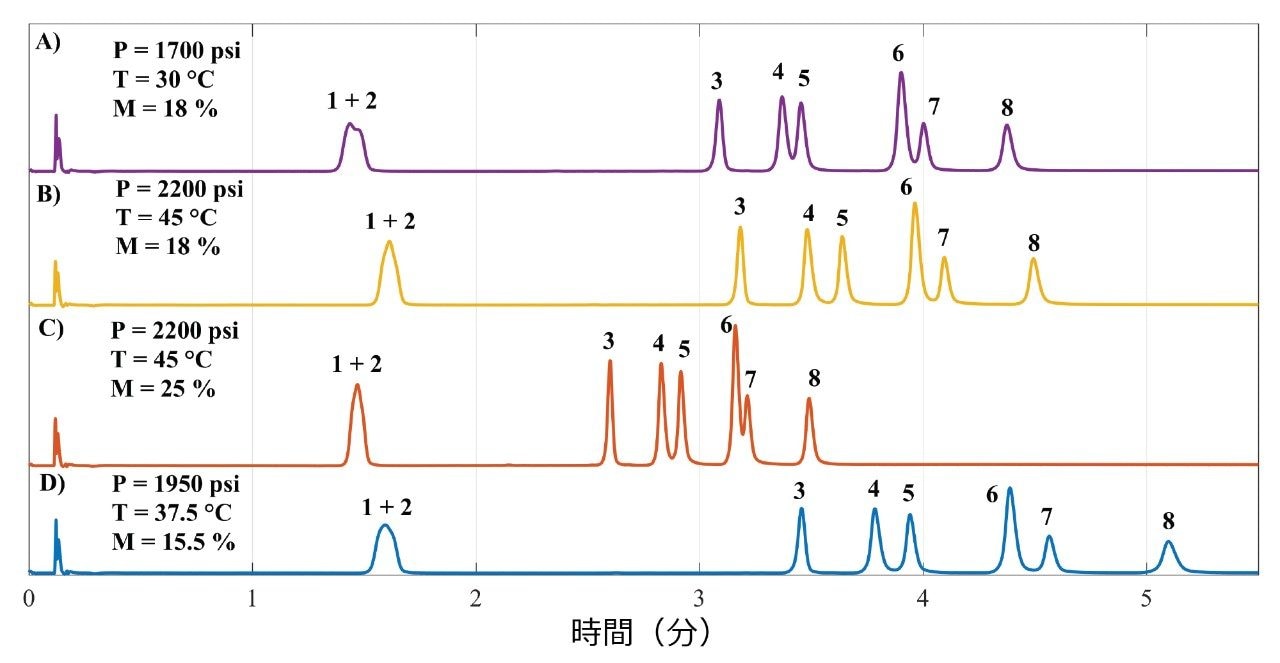
中心複合計画(Central Composite Design、CCD)を用いて、圧力、温度、モディファイヤーの比率といったパラメーターにより影響を受ける超臨界流体の密度を試験しました。そのため、6 つの軸点および中心点(5 回繰り返し測定)に沿って階乗 2³ を使用しました。この設定により、より少ない数の実験でナレッジスペースを拡張することができました19。図 2 に、19 のクロマトグラムのうち 4 つを示します。係数を変えることで異なるプロファイルが得られることがわかります。図 2A と 2B には、溶出プロファイルにわずかな差があり、図 2B に示す分離の方が優れています。いずれの場合も、グラジエント溶出終了時のモディファイヤーの成分は同じでしたが(18%)、図 2B の結果は、より高い圧力(2200 psi)および温度(45 ℃)の条件で得られたもので、これによって RSV/FLV および PRV/PTV の分離度が 1.5 より高くなりました。
更に、モディファイヤーの比率を 25% に設定した場合(図 2C)、溶出は速くなりましたが PRV および PTV が大きく重なりました。平均の圧力および温度がそれぞれ 1950 psi と37.5 ℃ で、モディファイヤーの比率が最低(15.5%)という別の条件では、クリティカルペアである RSV/FLV および PRV/PTV の分離度が 2.0 と最大になりました。図 2B および図 2D に示したクロマトグラムが最良の分離プロファイルであることがわかります。これらのクロマトグラムにより、低含量のモディファイヤーおよび高い圧力と温度で、頑健な分析法が得られることが示されました。最も影響が大きいパラメーターはモディファイヤーの比率で、保持時間が短縮し、移動相での化合物の溶解度が高くなりました。
EZT と ATV については、条件に関係なく十分な分離が得られましたが、SMV と LOV は、両者の類似性が高いために(前述したよう CH3 基が 1 つ)、どの条件でも分離できませんでした。そこで、クリティカルペアである RSV/FLV および PRV/PTV の分離を試験するに際して CCD に注目しました。モデル化のレスポンスとして分離係数(α)を使用しました。クリティカルペアの分離度 2.0 以上を達成するには、分離係数が 1.04 以上であることが必要です。分離度 2.0 以上を用いたのは、PTV と PRV の間の高さ比が 2:1 前後であったためです20。
興味深い点として、RSV および FLV の間の分離は温度の影響を強く受け(図 3A)、PRV および PTV の間の分離は、(赤い領域に向かって)主に圧力の影響を受ける(図 3B)ことも分かりました。これらの影響は、モディファイヤーの含量が低く、移動相の CO2 含量が高い場合(したがって流体の圧縮性が高い場合)に、更に顕著でした。言い換えると、流体パラメーターの内、温度と圧力の両方が、大きな影響を及ぼすことが予測されます。これにより、SFC 条件下で分離特性に影響を与えるパラメーターを試験する上において、多変数アプローチの使用の重要性が浮き彫りになります。
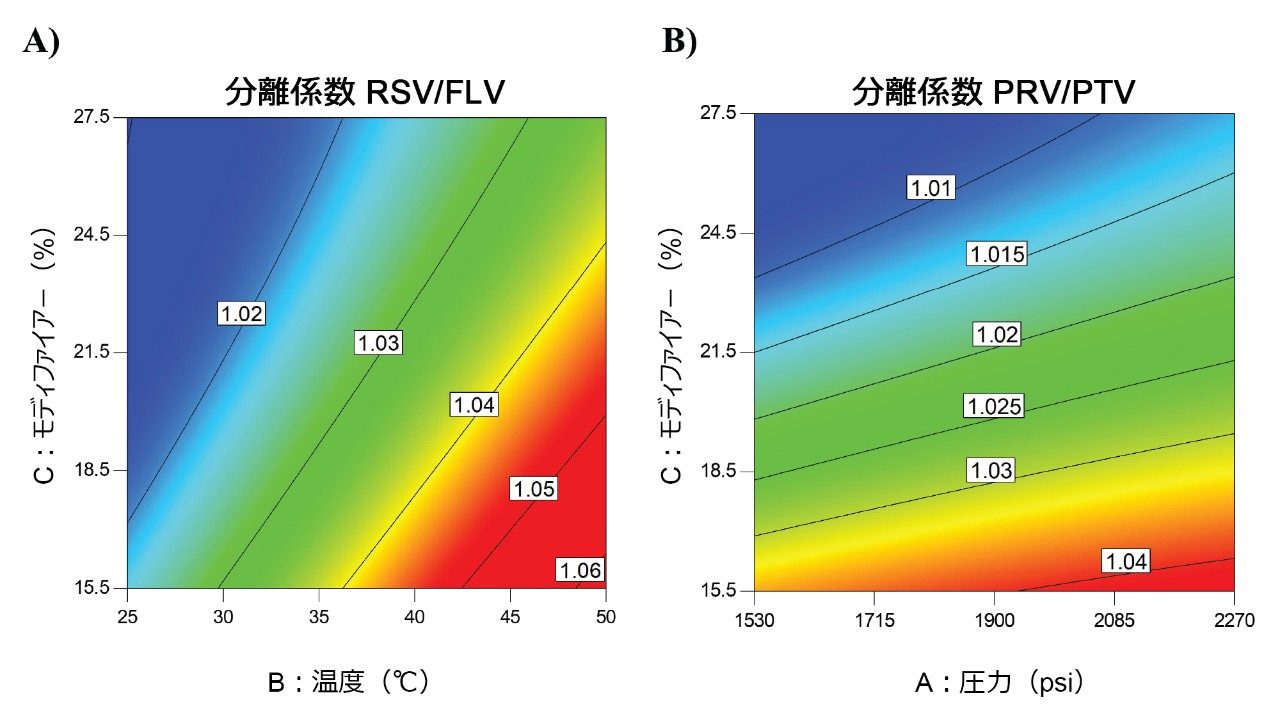
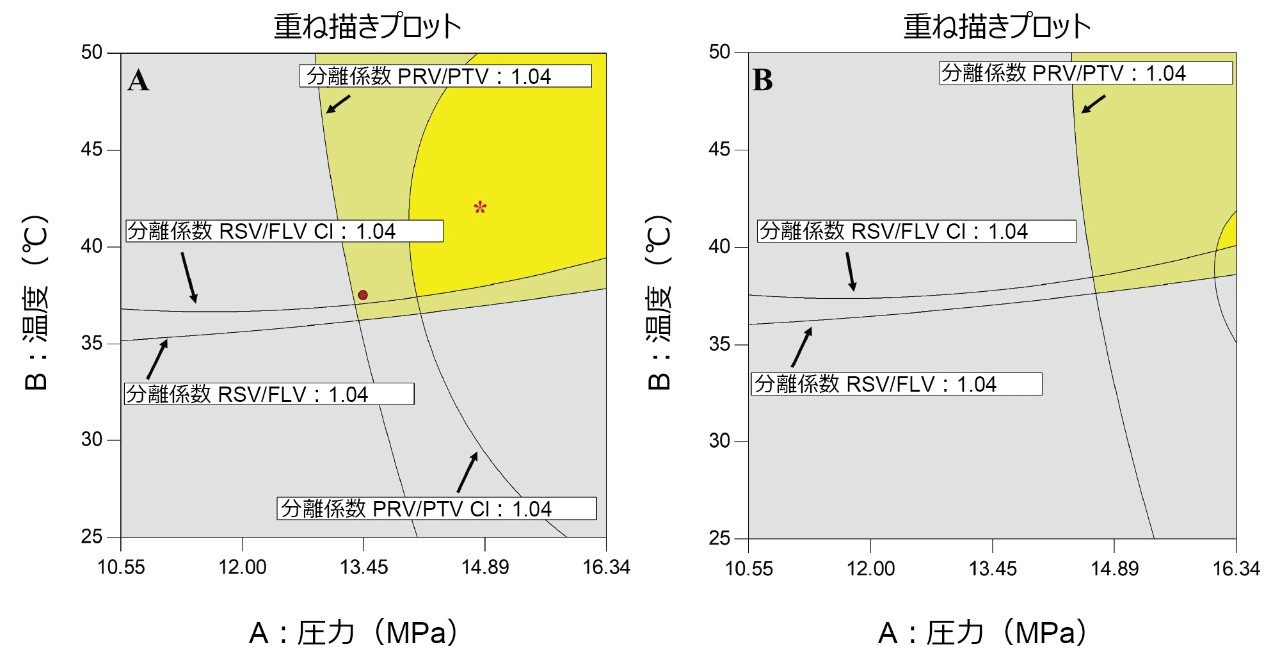
図 4 に、2 つのクリティカルペアの重ね描きプロットを、グラジエント終了時の有機モディファイヤーの含量 15.5%(図 4A)および 16.0%(図 4B)の場合について示します。明るい黄色の領域は、望ましいレスポンスの領域を示します。淡い黄色の領域は、予測が不確かな領域を示します(予測値平均についての信頼区間 95%)。グレーに色付けした領域は、許容されない係数設定を規定します。
バリデーション用に選択した分析法条件は、明るい黄色の領域内の温度 42 ℃、圧力 14.83 MPa、4 分間でモディファイヤー 5% ~ 15.5% のグラジエント溶出で、アスタリスク(*)で示しています。この分離を達成するための重要な変動要因は、図 4A と 4B の比較から分かるように、移動相中の有機モディファイヤーの比率です。これを把握することは、分析法開発時に CCD モデリングを使用することで始めて可能になりました。
最終段階では、分析時間を短縮する試みとして流速を評価しました。PTV および PRV のピークに、分離のわずかな改善が見られました(図 5)。これは、図 3 および図 4 に見られるように、流量の増加によって全体の圧力も高まり、結果として分離係数が高くなったためです。図 5 に、最初の流速(A、1.50 mL/分)および選択した流速(B、1.80 mL/分)でのクロマトグラムを示します。流速の上昇は、圧力限界(上限 41,4 MPa)によって妨げられると考えられます。
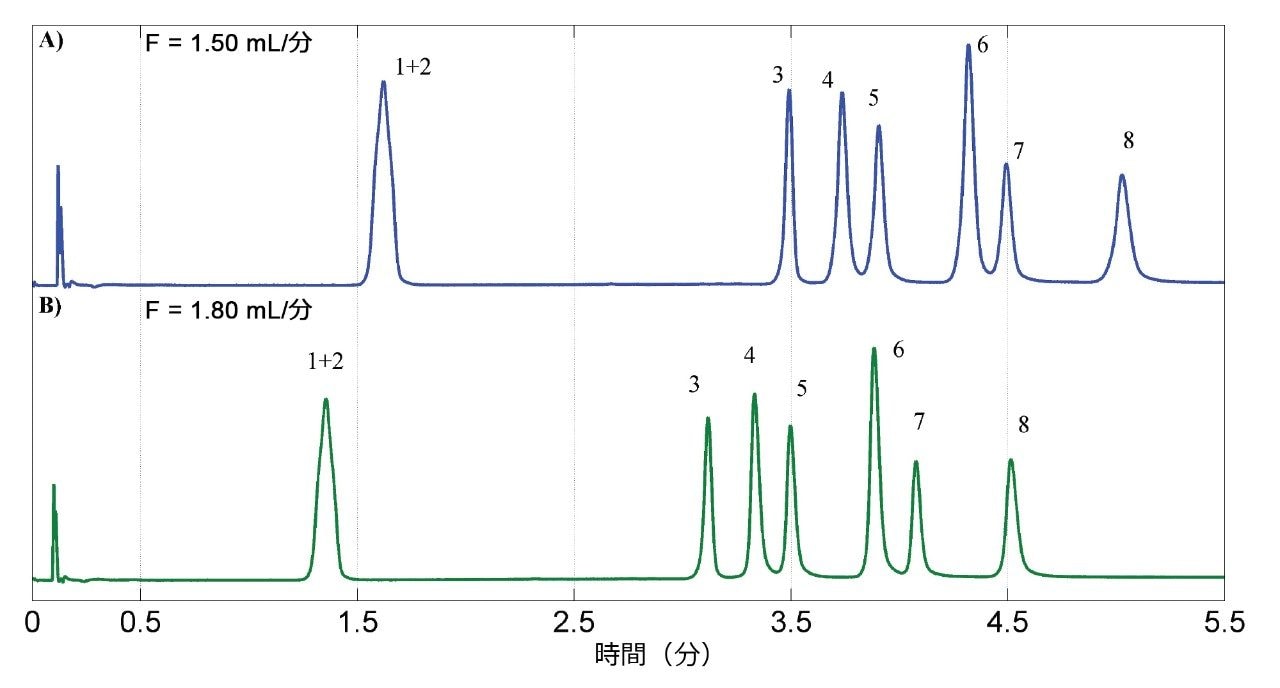
そのため、最適化した分析法の条件は、Torus 1-AA カラム、モディファイヤー EtOH:H2O 95:5(v/v)、流速 1.80 mL/分、圧力 14.83 MPa、温度 42 ℃、4 分間でのモディファイヤー 5% ~ 15.5% のグラジエント溶出の後、15.5% のアイソクラティックを更に 2 分間維持、注入量 1.0 µL、としました18。この分析法をバリデーションするために、以下のセクションで説明するように医薬品標準試料を使用しました。PDA 検出では、それぞれの分析種について 231 nm ~ 244 nm の範囲の特定の波長に設定しました。
分析法のバリデーション
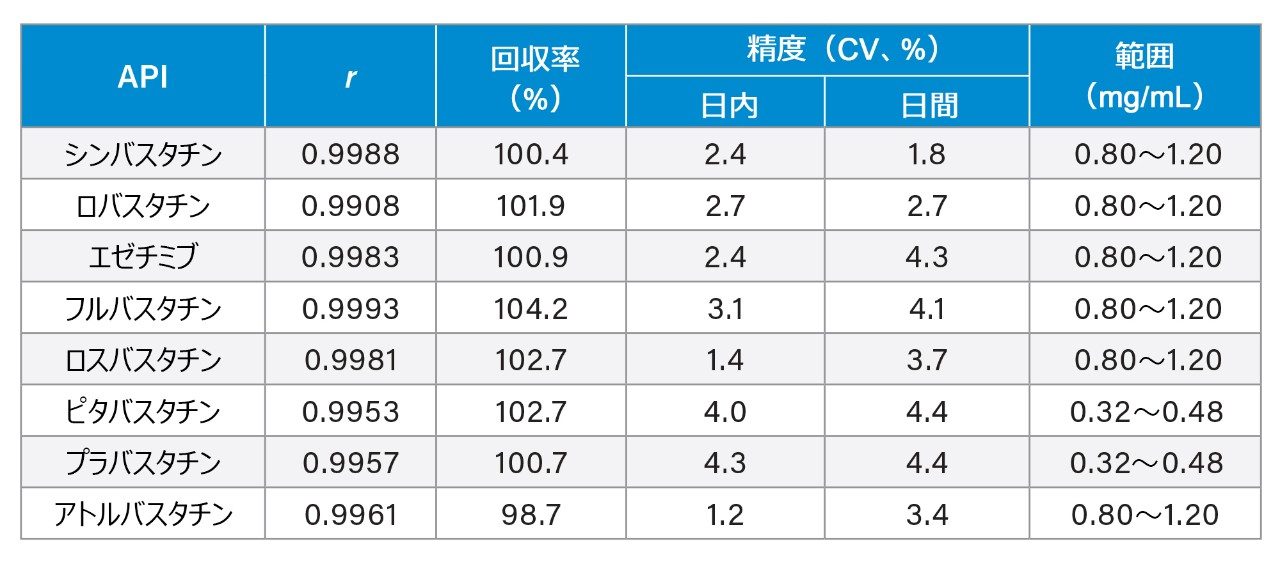
ブラジルの最新のガイドラインに従って分析法をバリデーションしました21。 最初のステップは分析法の選択性の評価で、ブランク(希釈剤のみ)、プラセボサンプル、および薬剤の標準溶液を注入しました。標準試料以外では、シグナルは観察されませんでした。その後、別々に調製した 3 つのストック溶液を 5 つの濃度に希釈して、検量線を作成しました。濃度範囲は 0.80 mg/mL ~ 1.20 mg/mL(PRV および PTV は 0.32 mg/mL ~ 0.48 mg/mL)でした。バリデーションのために評価した性能指数は、直線性、回収率に関する正確性、日内および日間の測定値のばらつきの係数に関する精度などです。この分析法は、最終製品の医薬品有効成分(API)のアッセイ用のみに提案したものであるため、検出限界および定量限界は評価しませんでした。
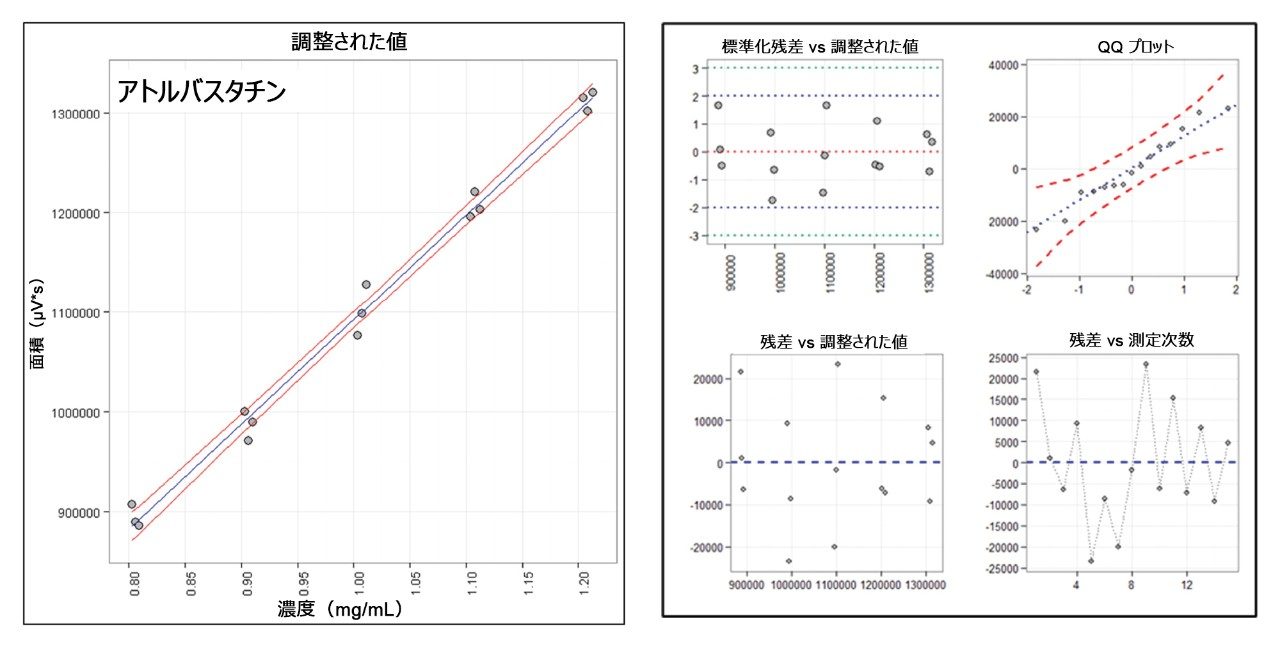
直線性は、ピアソンの相関係数(r)として測定し、結果は 0.99 以上でした。図 6A に一例として ATV の分析曲線、図 6B に対応する回帰の残差の分析を示します。ATV は、医薬品史上で販売量が最も多い製品リピトールを代表するものとして選択しました。表 1 に、得られた性能指数をまとめます。回収率および精度は、アッセイレベル 100% についてのみ示します。Horwitz の式に基づいて、使用した濃度での変動係数は 5.4% 以下と予測されます。回収率は 95% ~ 105% になると考えられます。試験した濃度すべてにおいてこの範囲内でした。
結論
この文書では、コレステロール低下薬の分析のための、最新の Torus 1-AA カラムと環境に優しい溶媒であるエタノールをより少量使用する、移動相添加剤が不要な ACQUITY UPC2 分析法 の開発およびバリデーションについて説明しました。連続 DoE アプローチは、圧力、温度、特にモディファイヤー比率などの要素が超臨界流体の溶出強度にどのように影響を及ぼし、分析種の保持および分離に影響するかを理解することで、分析法を最適化できる決定的なツールになります。
CCD の結果により、圧力、温度、有機モディファイヤーの比率が分析種の保持および分離に与える影響を理解でき、バリデーションのための頑健な作業ポイントが選択できます。有機モディファイヤーの比率が、望ましい分離を達成するために管理すべき重要な変動要因でした。最適化した分析法は、すべての分析種について、ブラジルの最新のガイドラインに従ってバリデーションしました。r が 0.990 を超えていることから正確性および精度が高いことがわかり、回収率は 95% ~ 105%、精度(日内および日間)は 4.4% 以下でした。11 種の市販製品のアッセイ分析から、アトルバスタチンを含む 1 種のみが USP 規格外であることが分かりました。そのため、これらの医薬品の分析において、ACQUITY UPC² は環境に優しい代替法であることが示されました。
参考文献
- Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region 2000–2016, Geneva, World Heal.Organ.2018.
- D.S. Kazi, J.M. Penko, K. Bibbins-Domingo, Statins for Primary Prevention of Cardiovascular Disease, Med.Clin.North Am.101, 2017.689–699.doi:10.1016/j.mcna.2017.03.001.
- S. Nodari, P. Rocca, A. Saporetti, L. Bettari, A.L. Foresti, E. Tanghetti, M. Metra, L. Dei Cas, The Combination of Ezetimibe and Statin: A New Treatment For Hypercholesterolemia, Heart Int.3, 2007.12. doi:10.4081/hi.2007.12.
- L. Nováková, D. Šatínský, P. Solich, HPLC Methods for the Determination of Simvastatin and Atorvastatin, Trends Anal.Chem. 27, 2008.352–367.doi:10.1016/j.trac.2008.01.013.
- M.I. Beludari, K.V. Prakash, G.K. Mohan, RP-HPLC Method for Simultaneous Estimation of Rosuvastatin and Ezetimibe from their Combination Tablet Dosage Form, Int.J. Chem.Anal.Sci. 4, 2013.205–209.doi:10.1016/j.ijcas.2013.04.006.
- M. Campos‐Lara, J.A. Mendoza‐Espinoza, Development of a Selective Extraction Method for Pravastatin Quantification in Tablets using HPLC with Ultraviolet Detection, J. Liq.Chromatogr.Relat.Technol. 31, 2008.619–623.doi:10.1080/10826070701815288.
- T.D. Silva, M. a Oliveira, R.B. de Oliveira, C.D. Vianna-Soares, Development and Validation of a Simple and Fast HPLC Method for Determination of Lovastatin, Pravastatin, and Simvastatin, J.Chromatogr.Sci. 2012.
- L. Nováková, A. Grand-Guillaume Perrenoud, I. Francois, C. West, E. Lesellier, D. Guillarme, Modern Analytical Supercritical Fluid Chromatography Using Columns Packed With Sub-2-μm Particles: A tutorial, Anal.Chim.Acta. 824, 2014 18–35.doi:10.1016/j.aca.2014.03.034.
- L. Ferey, A. Raimbault, I. Rivals, K. Gaudin, UHPLC Method for Multiproduct Pharmaceutical Analysis by Quality-by-Design, J. Pharm.Biomed.Anal. 148 (2018) 361–368.doi:10.1016/j.jpba.2017.10.014.
- A. Grand-Guillaume Perrenoud, J.L. Veuthey, D. Guillarme, Comparison of Ultra-High Performance Supercritical Fluid Chromatography and Ultra-High Performance Liquid Chromatography for the Analysis of Pharmaceutical Compounds, J. Chromatogr.A. 1266, 2012.158–167.doi:10.1016/j.chroma.2012.10.005.
- A. Tarafder, Metamorphosis of Supercritical Fluid Chromatography to SFC: An Overview, Trends Anal.Chem. 81, (2016) 3–10.doi:10.1016/j.trac.2016.01.002.
- D.B. Hibbert, Experimental Design in Chromatography: A Tutorial Review, J. Chromatogr.B. 910, 2012.2–13.doi:10.1016/j.jchromb.2012.01.020.
- A. Dispas, P. Lebrun, B. Andri, E. Rozet, P. Hubert, Robust Method Optimization Strategy – A Useful Tool for Method Transfer: The Case of SFC, J. Pharm.Biomed.Anal.88, 2014.519–524.doi:10.1016/j.jpba.2013.09.030.
- V. Abrahamsson, M. Sandahl, Impact of Injection Solvents on Efficiency in Supercritical Fluid Chromatography, J. Chromatogr.A. 1306, 2013.80–88.
- E. Lesellier, C. West, The many faces of packed column supercritical fluid chromatography - A critical review, J. Chromatogr.A. 1382, 2015.2–46.doi:10.1016/j.chroma.2014.12.083.
- C. West, E. Lemasson, S. Bertin, P. Hennig, E. Lesellier, An Improved Classification of Stationary Phases for Ultra-High Performance Supercritical Fluid Chromatography, J. Chrtitomatography A. 1440, 2016.212–228.doi:10.1016/j.chroma.2016.02.052.
- C. West, E. Lesellier, Characterisation of Stationary Phases in Subcritical Fluid Chromatography With the Solvation Parameter Model: III.Polar Stationary Phases, J. Chromatogr.A. 1110, 2006.200–213.doi:10.1016/j.chroma.2006.01.109.
- I. M. Santana, I. C. S. F. Jardim, M. C. Breitkreitz, Sequential Design of Experiments Approach for the Multiproduct Analysis of Cholesterol-Lowering Drugs by Ultra-High-Performance Supercritical Fluid Chromatography, J. Sep. Sci., 43, 2020, 4234–4242.doi: 10.1002/jssc.202000702.
- P.K. Sahu, N.R. Ramisetti, T. Cecchi, S. Swain, C.S. Patro, J. Panda, An Overview of Experimental Designs in HPLC Method Development and Validation, J. Pharm.Biomed.Anal. 147, 2018.590–611.doi:10.1016/j.jpba.2017.05.006.
- L.R. Snyder, J.J. Kirkland, J.L. Glajch, Practical HPLC Method Development, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1997.doi:10.1002/9781118592014.
- Agência Nacional de Vigilância Sanitária, Resolução RDC 166.2017.
720007390JA、2021 年 11 月