In the pharmaceutical industry, rapid preparative isolation is required for a number of reasons. These range from the use of chromatographic markers for the purpose of structure elucidation, for determination of relative response factors, and for reference standards. Current regulatory requirements on the control of impurities and degradation products in drug substances and drug product, dictate that for impurities at levels greater than 0.1%, unambiguous characterization is required.1 Modern, sophisticated, spectral hyphenated techniques, such as liquid chromatography mass spectrometry (LC-MS), liquid chromatography mass spectrometry-mass spectrometry (LC-MS/MS), and liquid chromatography nuclear magnetic resonance (LC-NMR), are already being extensively used by the industry, but in many instances, these techniques in isolation are not successful and an extra purification step is required to isolate the impurities for subsequent unambiguous characterization.2
Supercritical fluid chromatography (SFC) is a normal-phase chromatographic technique which found its place early in its development with chiral separations. In recent years, the improved quality of available hardware has led to a wider uptake of the technology for achiral separation applications, and has replaced reverse phase liquid chromatography (RPLC) separation techniques for the resolution of closely related species within the pharmaceutical and life science industries.3
The use of a super critical gas in chromatography, typically carbon dioxide due to the easily achievable conditions required to reach its critical point, (74 bar pressure and >32 °C temperature) offers several key benefits. Once above the critical point the liquid and gas phases are no longer distinct, meaning the super critical phase has solubilizing properties which are similar to a liquid with gas-like diffusivity. This translates chromatographically to lower back-pressures, allowing the use of higher flow rates, resulting in quicker analysis and more efficient chromatographic separations. SFC has also been praised as a ‘green’ separation technique eliminating the need for organic solvents such as heptane and hexane. Typical co-solvents used in SFC are alcohols (methanol, ethanol, or isopropanol) which are seen as a preferred solvent of choice because they are recognized by most as green chemistry initiatives.
For preparative and semi-preparative applications, the use of SFC allows the majority of the mobile phase to be a gas under normal temperatures and pressures, meaning that the resulting fractions are lower volume organic solutions. This reduces the time and cost associated with solvent evaporation. The use of SFC for achiral applications also avoids the use of pH modifiers, ion pair reagents, and buffers, all of which can be left as residues in final compounds or may interact with chemically sensitive analytes on concentration and dry down.
Traditionally dominated by reverse phase liquid chromatography (RPLC), there is an increasing trend toward using supercritical fluid chromatography (SFC) to replace RPLC for purifications from the semi-preparative scale up to the kilogram scale.4,5
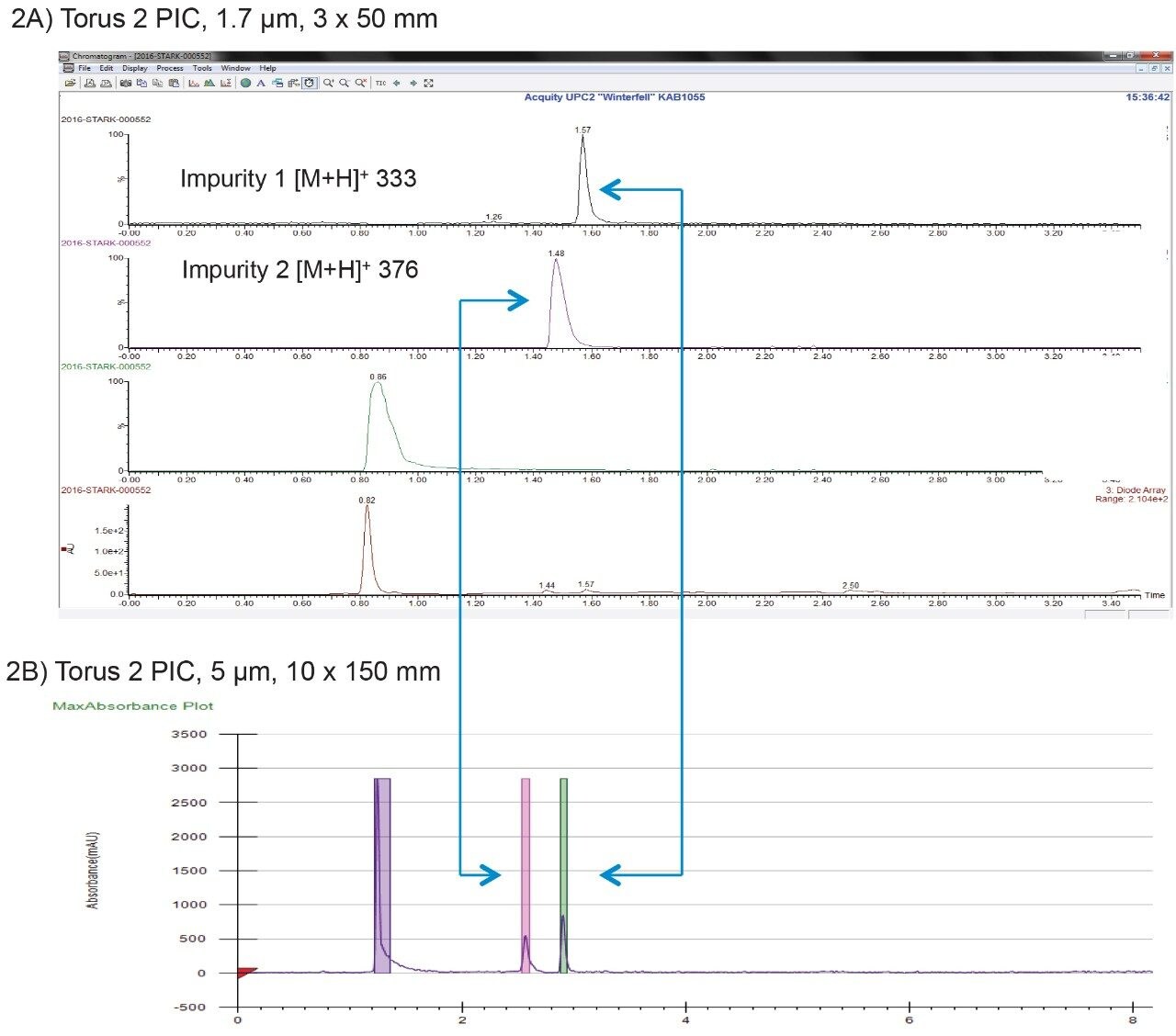
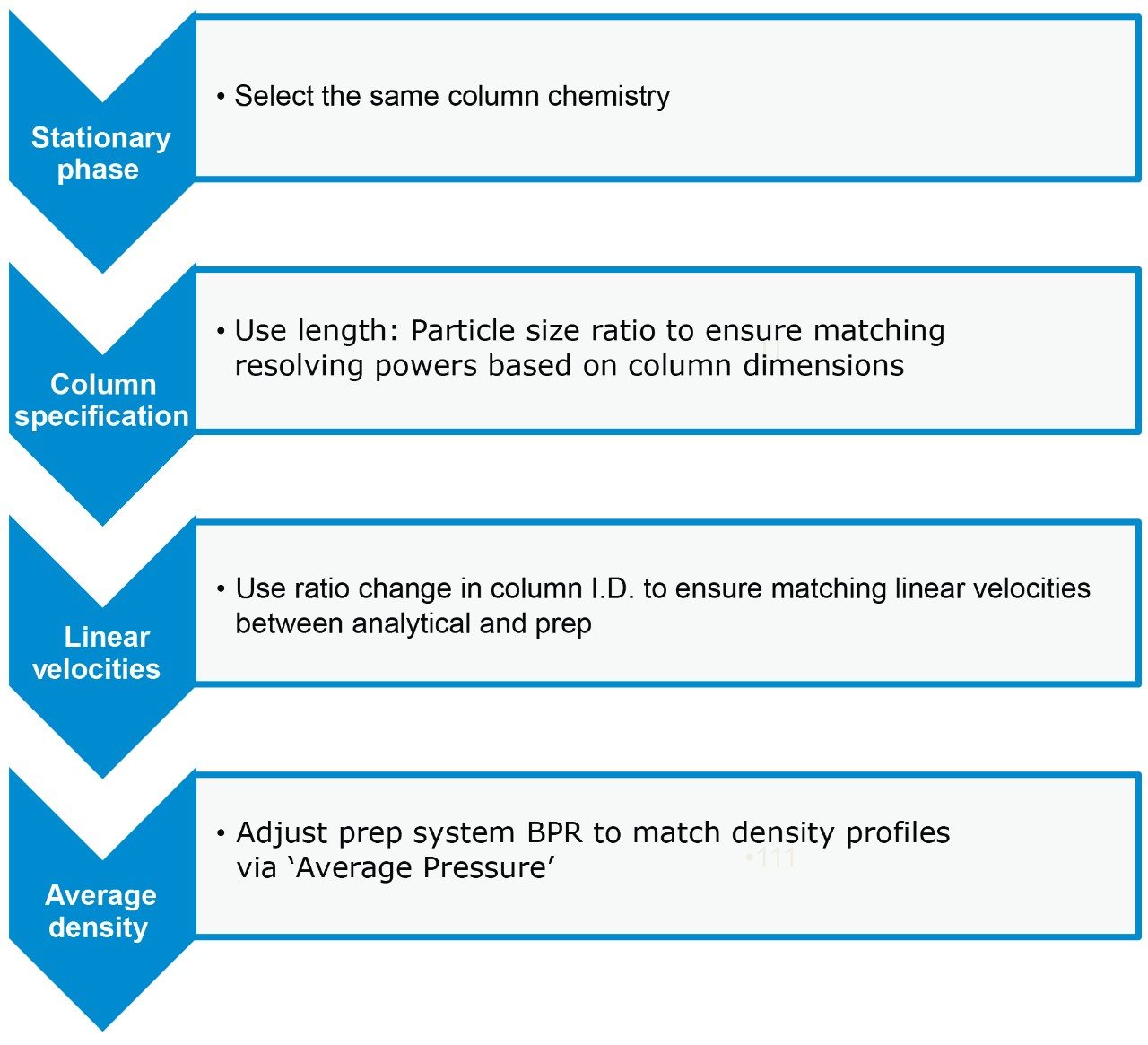
In this application note we compare the use of both preparative RPLC and SFC techniques, discussing the scaling process from the analytical to preparative columns while illustrating the efficiency gains when using preparative SFC to isolate the impurities.