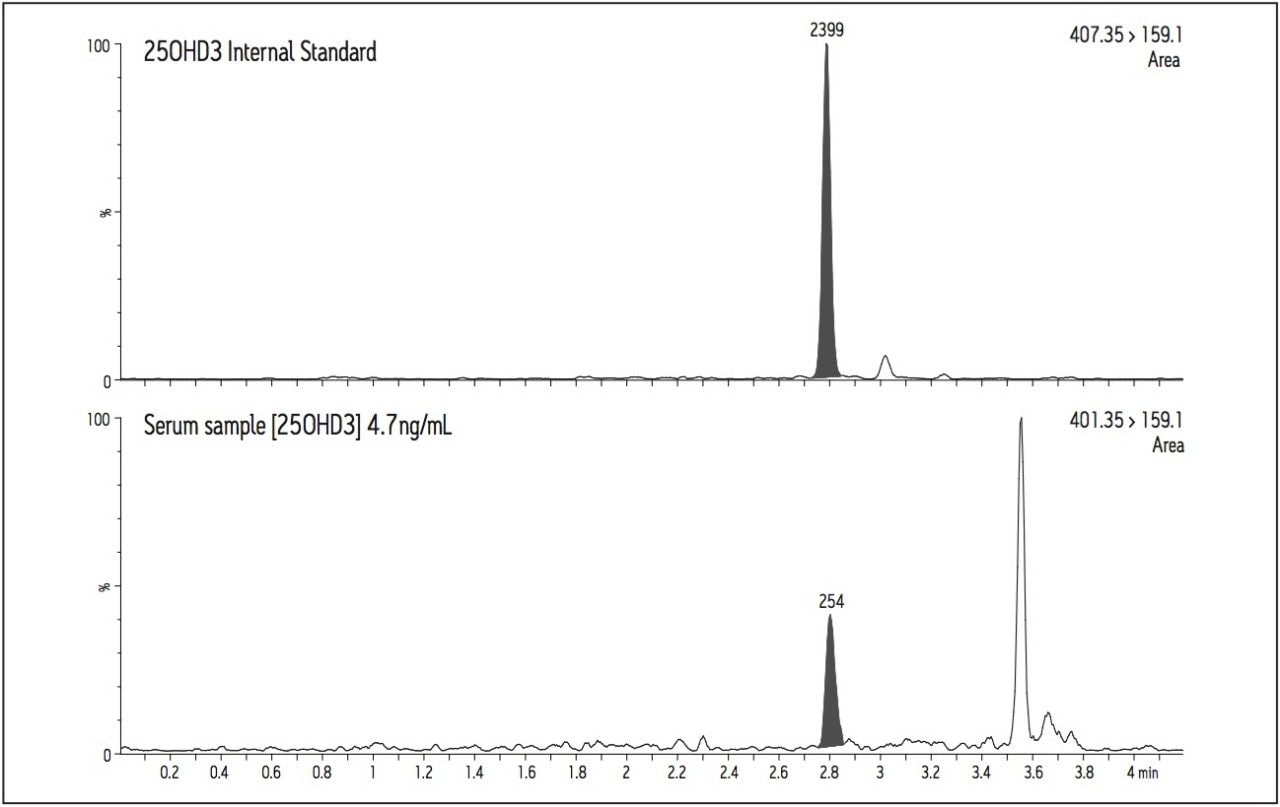
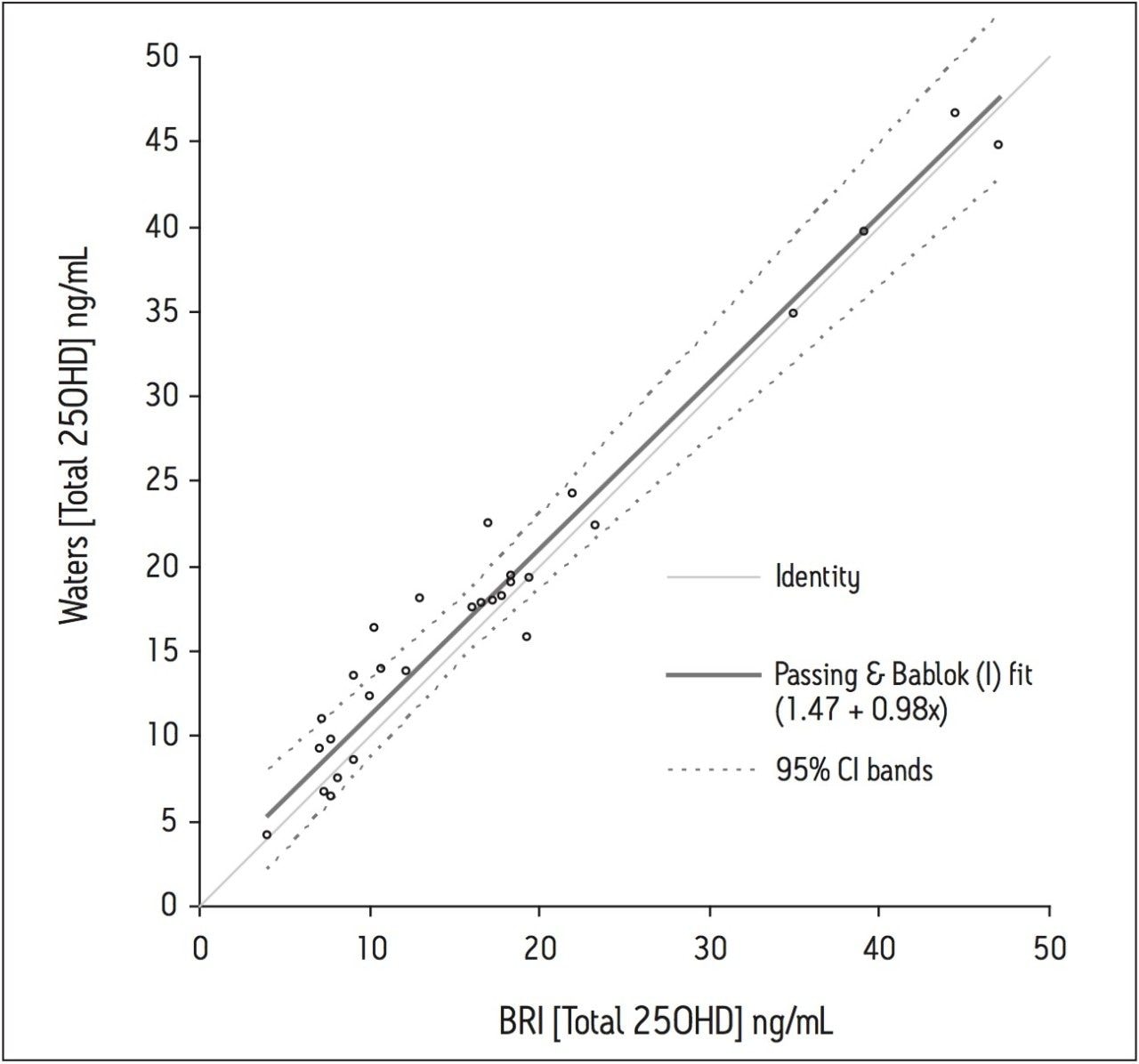
The demand for serum 25-hydroxyvitamin D, 25(OH)D, measurements has increased dramatically in recent years. While the role of vitamin D in bone metabolism is well established, comparatively little is known about its role in other diseases, although recent retrospective analysis of clinical trials data suggests possible links between vitamin D deficiency and a variety of diseases. Currently, considerable time, effort and funds are being applied to randomized, prospective clinical trials that aim to better define the link between vitamin D status and a variety of diseases, such as cancers, multiple sclerosis, heart disease and diabetes.1,2 Thus, an analytically accurate and precise measurement procedure having automated sample preparation is required to process the large number of samples from these clinical trials.
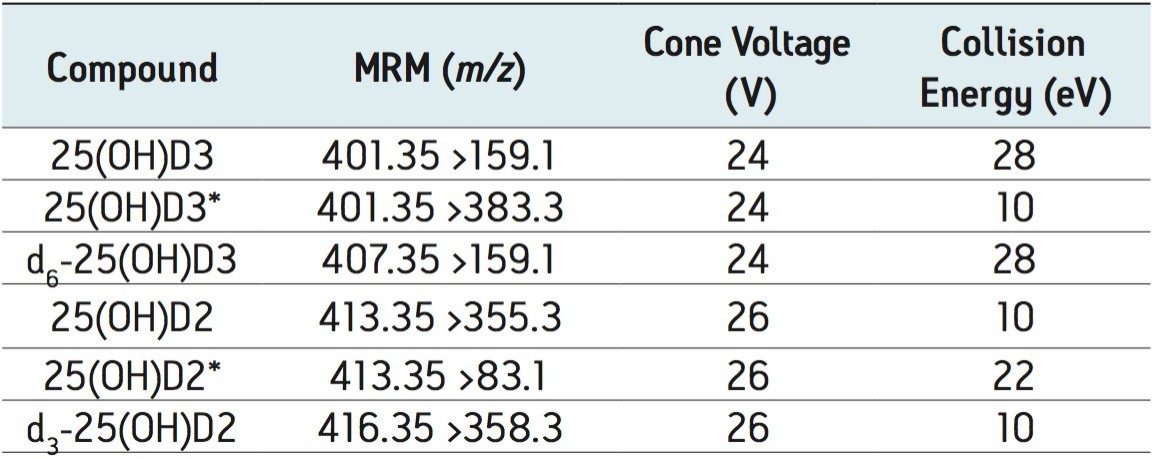
Vitamin D is available in two forms: the plant-derived vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol), formed upon exposure of the skin to ultraviolet radiation. The accepted indicator of vitamin D status, total 25(OH)D [that is, the sum of 25(OH)D2 and 25(OH)D3] has been a challenge to measure accurately because the antibodies used in some immunoassays do not have 100% co-specificity for both 25(OH)D2 and 25(OH)D3. In fact, some immunoassays may under-report the total 25(OH)D level in samples from subjects receiving vitamin D2 supplementation.3 Therefore, many clinical research laboratories have now adopted LC-MS/MS based methods for measuring total 25(OH)D; allowing independent quantification of 25(OH)D2 and 25(OH)D3.
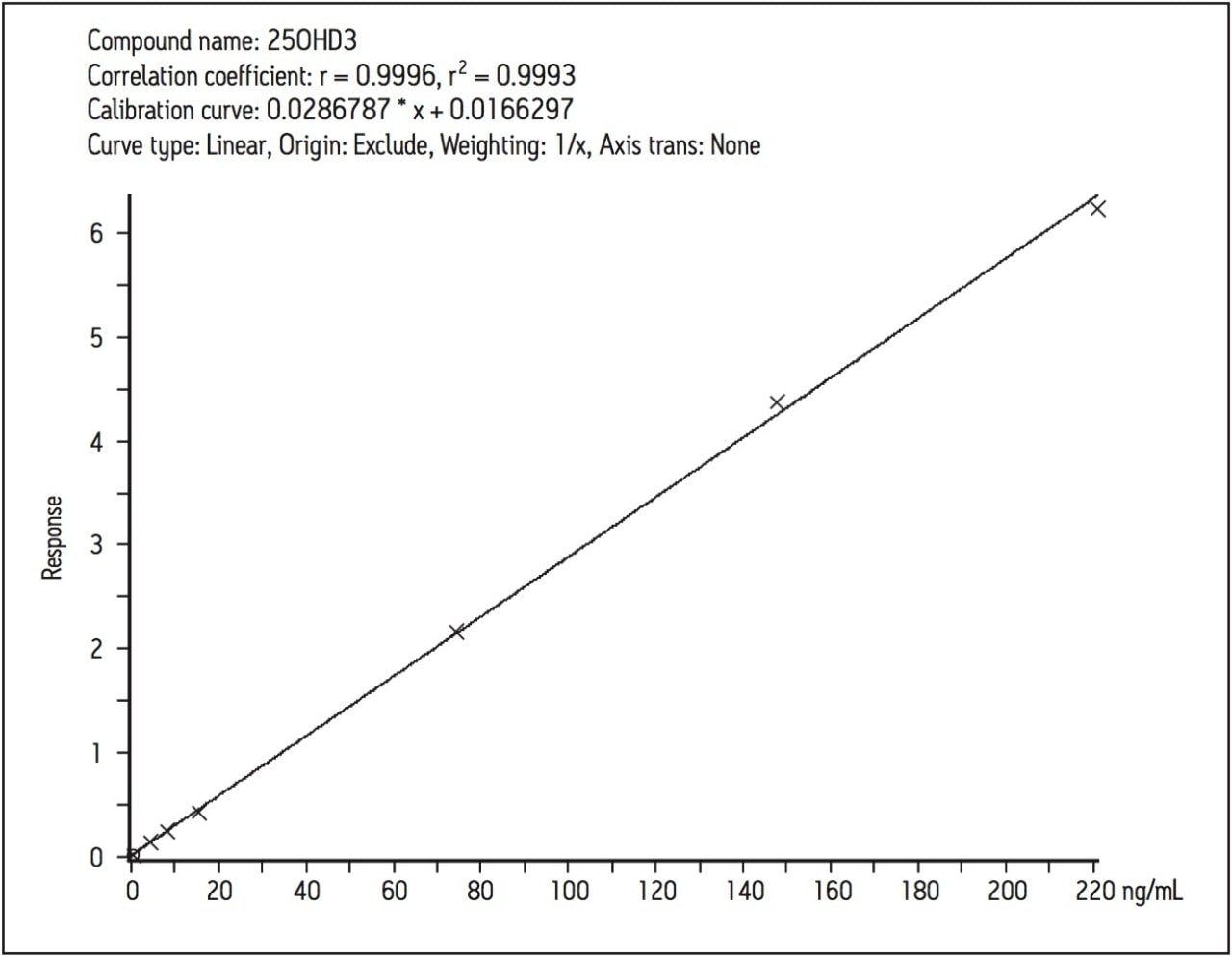
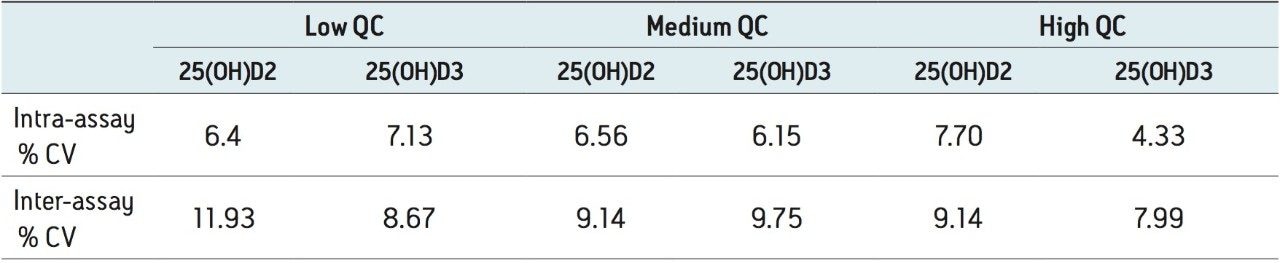
The analysis of 25(OH)D by LC-MS/MS requires sample pre-treatment to release it from the vitamin D binding protein and to minimize matrix effects. However, these steps are time-consuming and sample transfers may be subject to human error. This application note describes a routine UPLC-MS/MS procedure for clinical research for measuring 25(OH)D utilizing an offline automated sample pretreatment with sample tracking to process samples from the primary tube to final results.