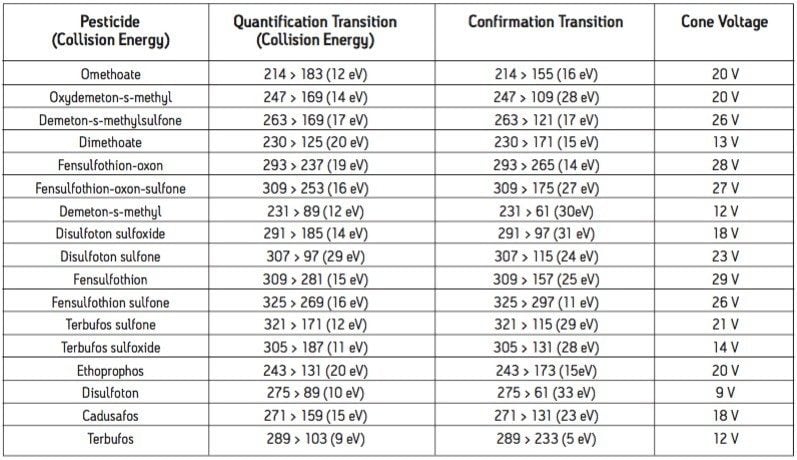
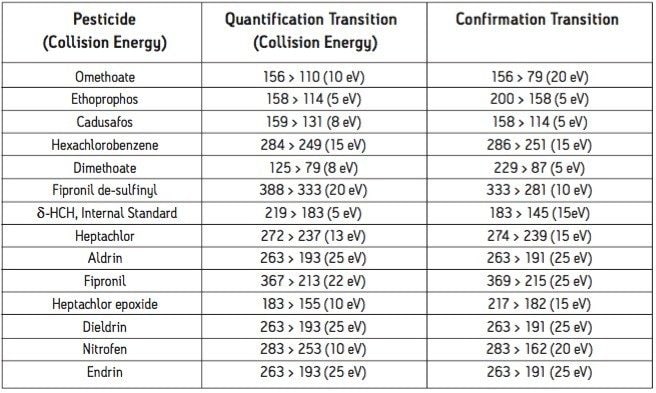
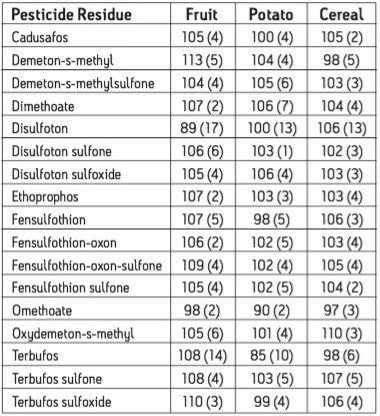
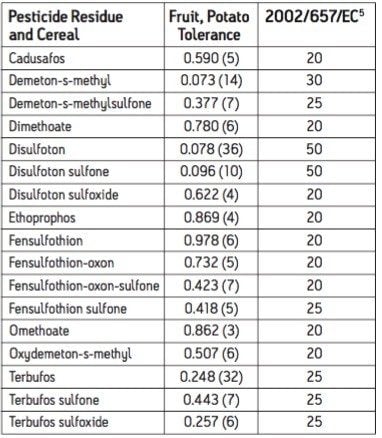
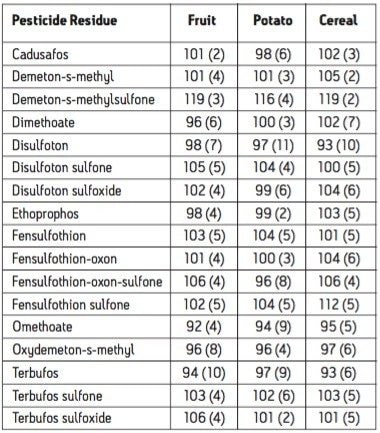
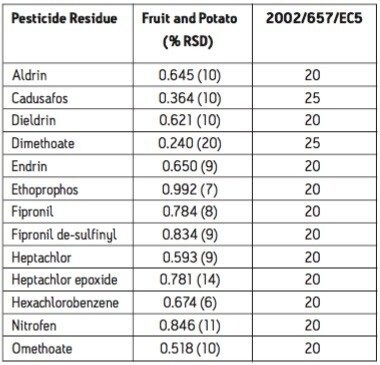
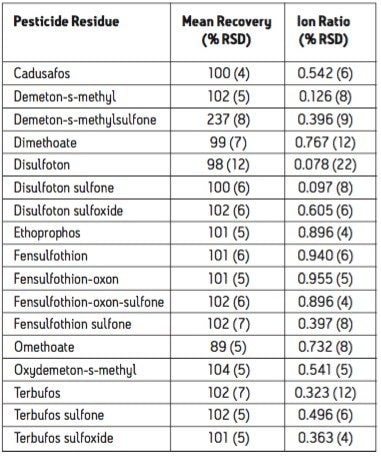
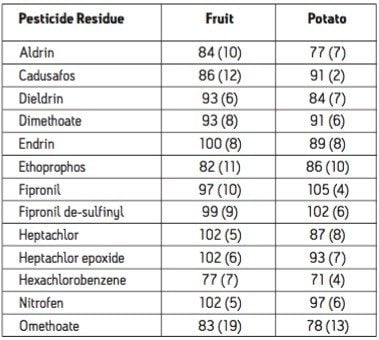
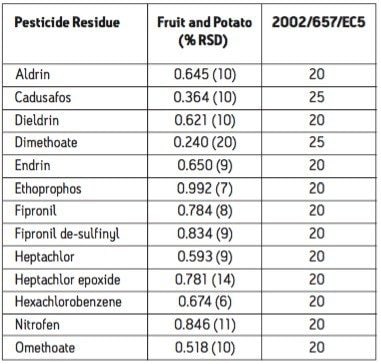
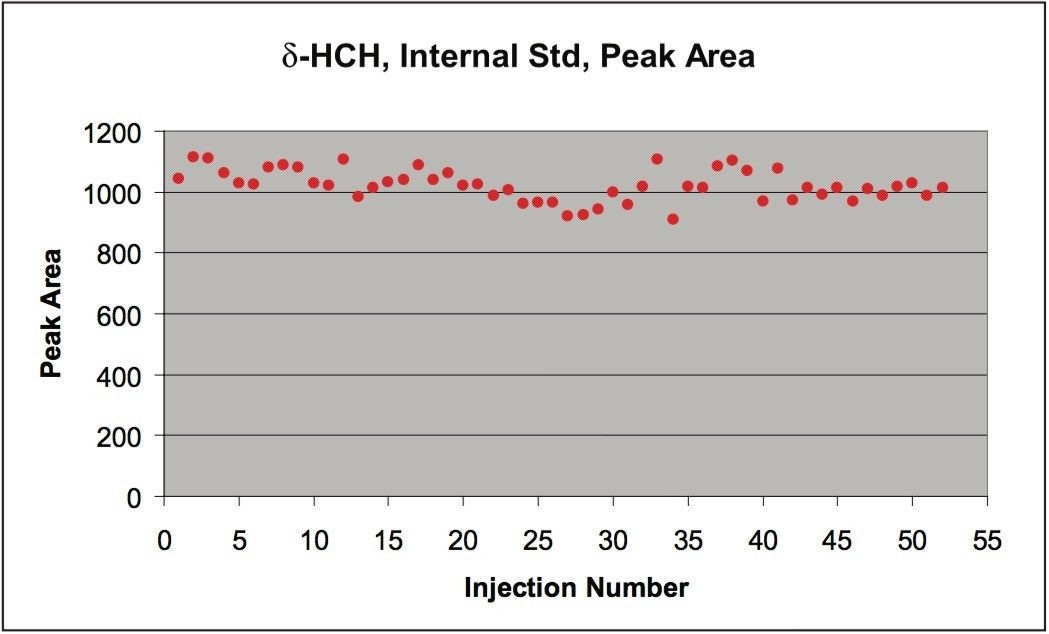
The European Union Baby Food Directive 2003/13/EC1 designates pesticides as prohibited, in which case they are considered not to have been used if their residue does not exceed 0.003 mg/kg or have maximum residue limits (MRLs) set between 0.004–0.008 mg/kg. Seven pesticides and nine transformation products (e.g. metabolites) listed in the Directive are suitable for LC-MS analysis while nine pesticides and three transformation products are amenable to GC-MS. The other pesticides specified in the Directive, because of their physicochemical properties, must be analyzed by single residue methods. Dimethoate was only included in the compound list as a possible precursor for omethoate.
To be able to enforce the Directive, laboratories require multiresidue methods with lower limits of detection (LOD) than those currently available. This necessitates improvements in the extraction, clean up, separation, and detection of pesticides in baby food samples. An extraction, with acetonitrile, followed by dispersive SPE clean up was reported for the analysis of a wide range of pesticides in fruits and vegetables2 and fatty samples.3 Acetonitrile extracts are suitable for direct analysis using LC-MS/MS, and by GC-MS/MS using programable temperature vaporization (PTV) in solvent vent mode.
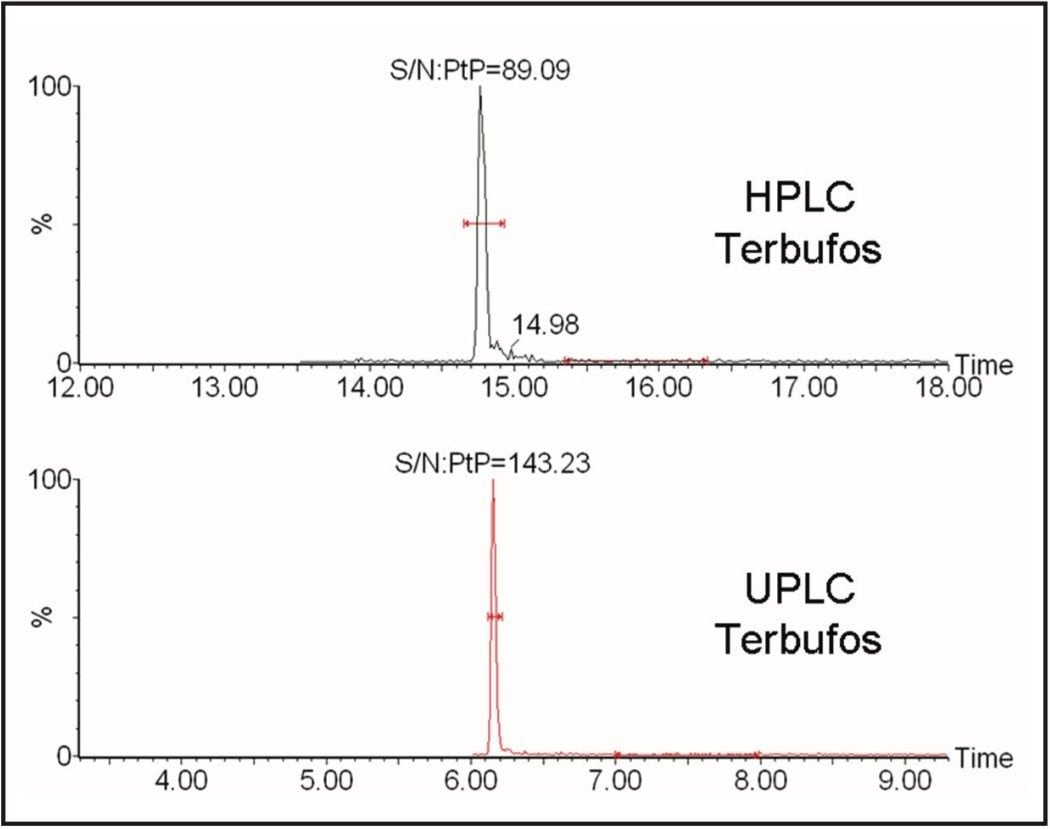
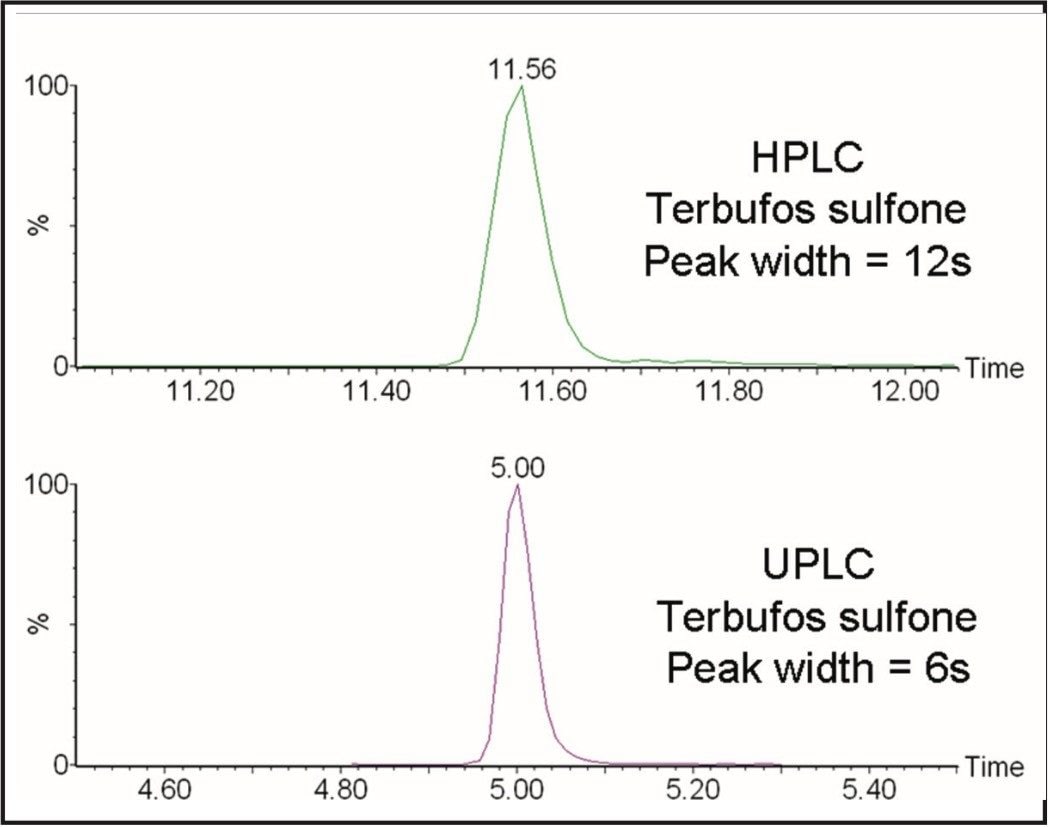
GC and HPLC have both been widely used in laboratories for the analysis of pesticide residues in food. The Waters ACQUITY UltraPerformance LC (UPLC)4 has the potential to provide shorter run times, greater sensitivity and better chromatographic resolution than established HPLC methods.