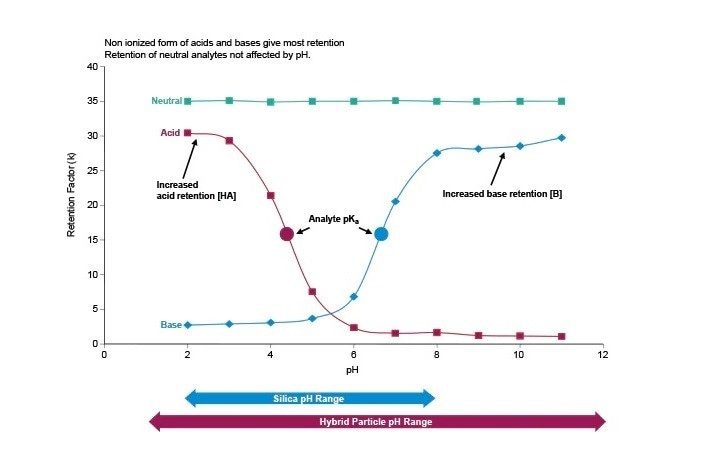
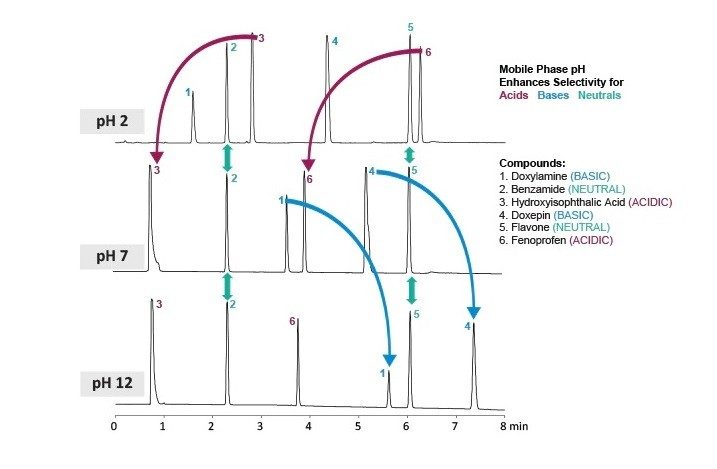
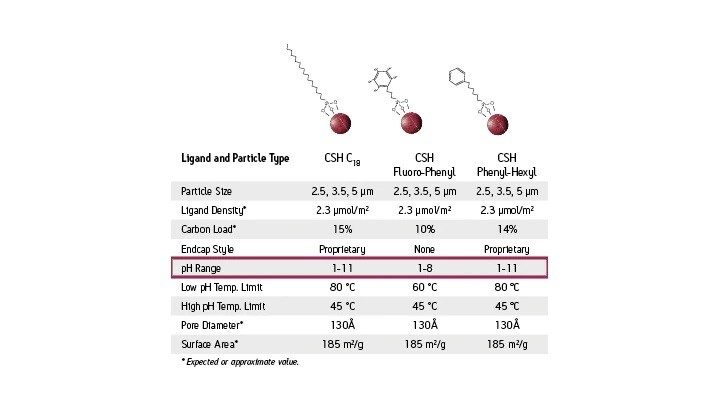
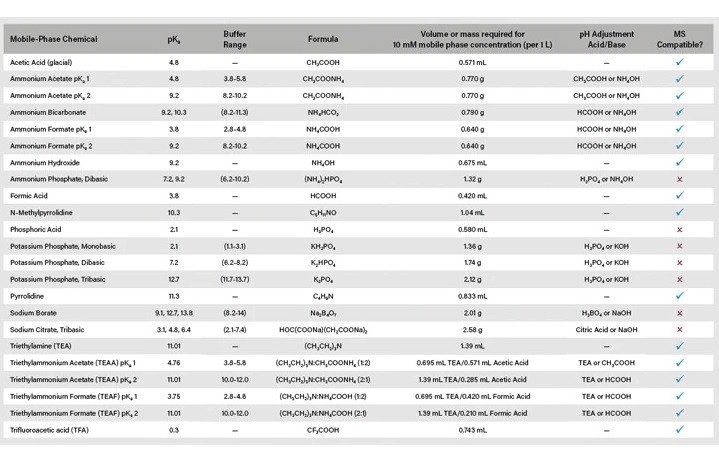
Temperatura
Poiché temperature diverse possono influire sulla selettività, riscaldare la colonna è uno strumento utile per ottimizzare la separazione di un campione specifico. La viscosità della fase mobile e la pressione totale del sistema si riducono con l’aumento della temperatura, il che si traduce in una minore sollecitazione del sistema, dei raccordi e della colonna, fino a incrementare la robustezza del funzionamento. Anche la temperatura può ridurre i tempi di ritenzione e modificare la selettività della separazione. È difficile prevedere se le variazioni di temperatura incrementeranno o diminuiranno la risoluzione dei picchi, pertanto l’utilità è specifica per ogni separazione specifica.
Mentre il controllo della temperatura nella cromatografia su piccola scala è di routine, la temperatura è impiegata raramente come parametro per manipolare la cromatografia destinata alla purificazione su scala preparativa. In primo luogo, colonne di grande diametro possono essere difficili da riscaldare uniformemente dall’esterno. In secondo luogo, le separazioni ad alta velocità di flusso usate su larga scala tendono a rimanere alla temperatura del solvente in ingresso. I mantelli elettrici e i forni colonna, sebbene soddisfacenti per separazioni su piccola scala, non sono in grado di riscaldare una colonna di grandi dimensioni in modo uniforme lungo l’intero diametro della colonna. Di conseguenza, sul diametro della colonna si formano gradienti di temperatura che influiscono negativamente sulla cromatografia.
Se il controllo della temperatura è necessario per mantenere la solubilità del campione, è possibile superare i gradienti di temperatura collocando la colonna preparativa in un bagnomaria riscaldato con un tratto di tubo collegato alla testa della colonna. Il tratto di tubo funge da preriscaldatore per equilibrare il solvente in ingresso alla temperatura desiderata. La cromatografia destinata a uno scale-up futuro viene sviluppata al meglio in condizioni ambiente e il controllo della temperatura è utilizzato come ultima risorsa.