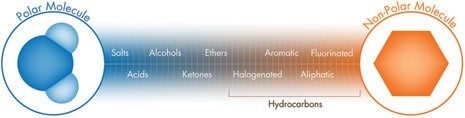
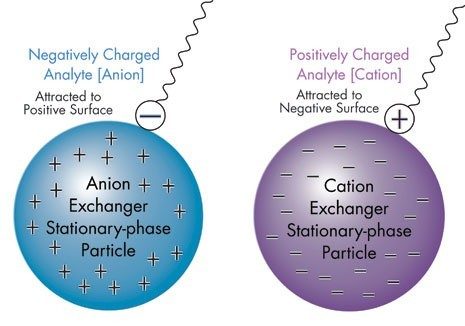
Quando gli scambiatori ionici deboli vengono neutralizzati, possono trattenere e separare le specie tramite interazioni idrofobiche (in fase inversa) o idrofile (in fase normale); in questi casi, l’intensità di eluizione è determinata dalla polarità della fase mobile (Figura R-1). Pertanto, è possibile utilizzare scambiatori ionici deboli per le separazioni in modalità mista (separazioni basate sia sulla polarità che sulla carica).
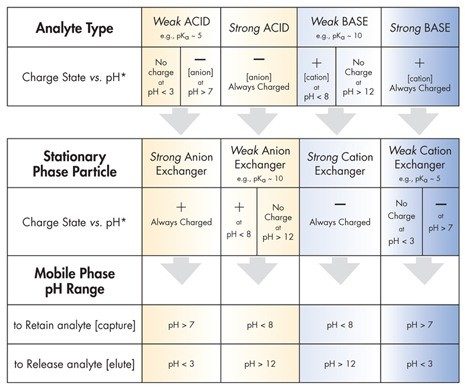
La Tabella D illustra le linee guida per le principali categorie di scambio ionico. Per esempio, per trattenere un analita altamente basico (sempre con carica positiva), utilizzare una particella in fase stazionaria a scambio cationico debole con pH >7; questo assicura una superficie delle particelle con carica negativa. Per rilasciare o eluire la base forte, abbassare il pH della fase mobile al di sotto di 3; in questo modo si rimuove la carica superficiale e si disattiva il meccanismo di ritenzione a scambio ionico.
Si noti che il pKa è il valore di pH in corrispondenza del quale il 50% del gruppo funzionale risulta ionizzato e il 50% risulta neutro. Per garantire una superficie dell’analita o delle particelle essenzialmente neutra, o completamente carica, il pH deve essere regolato a un valore di almeno 2 unità oltre il pKa, come opportuno (fare riferimento alla Tabella D).
Non utilizzare uno scambiatore cationico forte per trattenere una base forte; entrambi rimangono carichi e fortemente attratti l’uno dall’altro, rendendo quasi impossibile l’eluizione della base. Questa può essere rimossa solo inondando lo scambiatore cationico forte con una base alternativa che presenti una capacità di ritenzione ancora maggiore e sposti il composto di interesse vincendo la competizione per i siti di scambio ionico attivi. Questo approccio è raramente pratico o sicuro nell’ambito della HPLC e della SPE. [È pericoloso lavorare con acidi e basi molto forti che possono, inoltre, essere corrosivi per i materiali di costruzione utilizzati nei sistemi HPLC!]