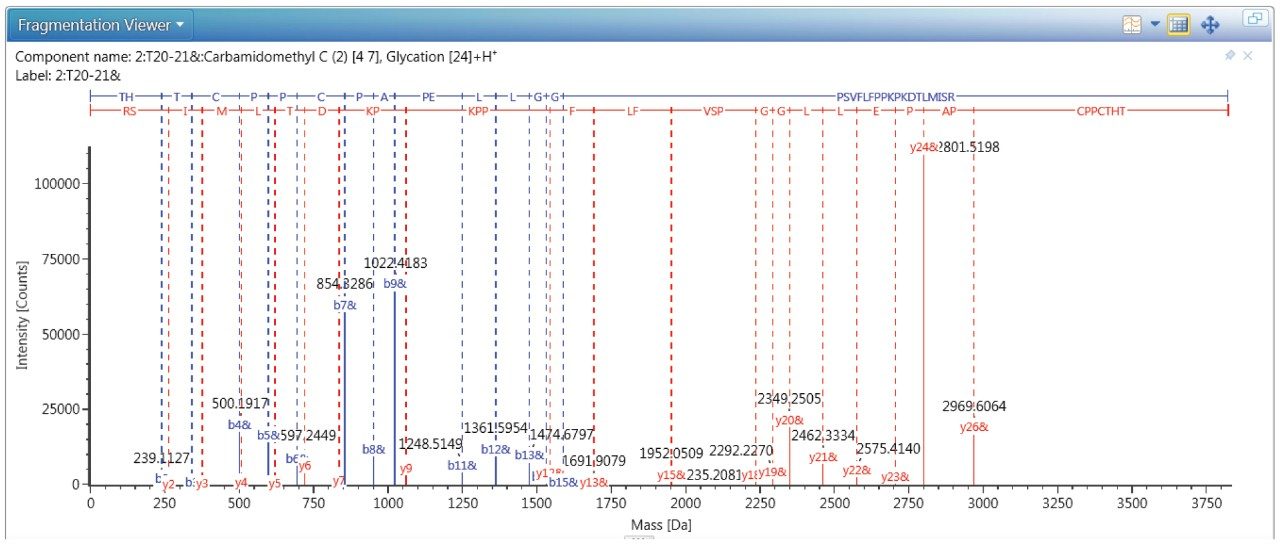
Low level PTM characterizations: example on glycation site identification
With naturally low abundance in NIST mAbs, glycated peptides usually show very little fragmentation across the peptide bonds in an MSE experiment. In this application note, we demonstrate an optimized DDA method to identify the glycated peptides using NIST mAb RM 8671 due to its high glycation level.
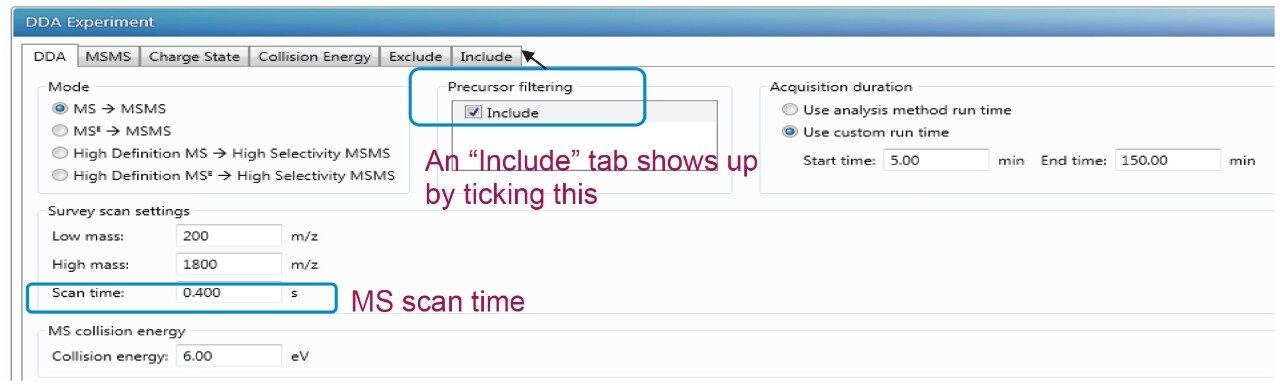
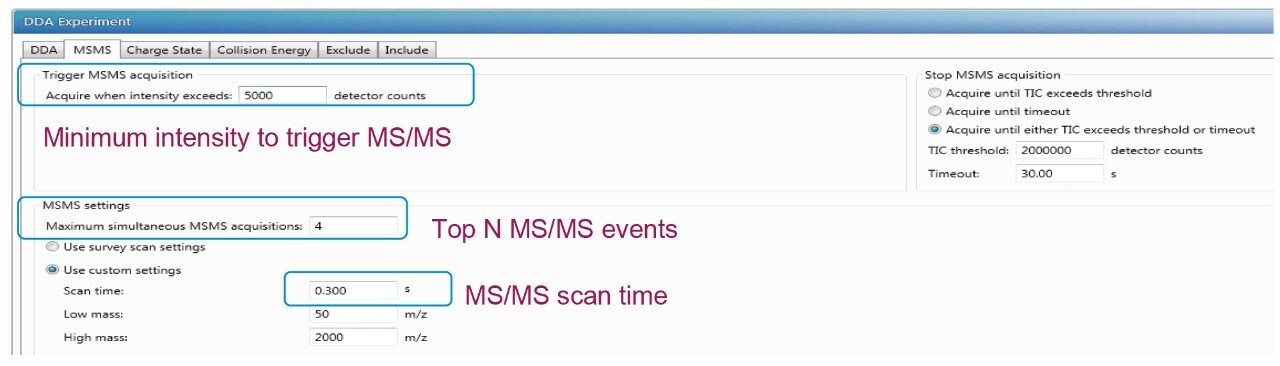
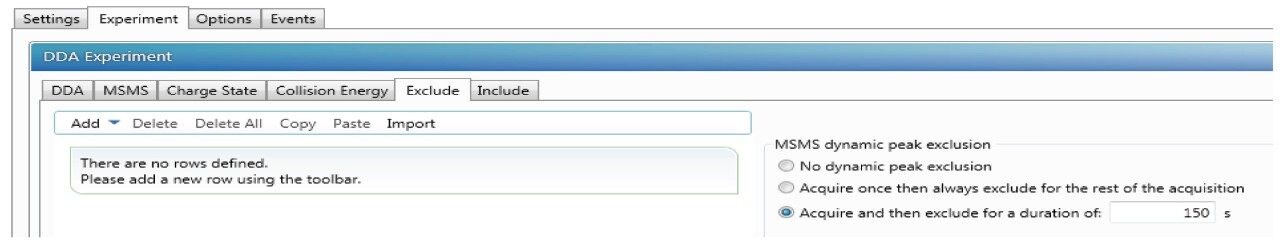
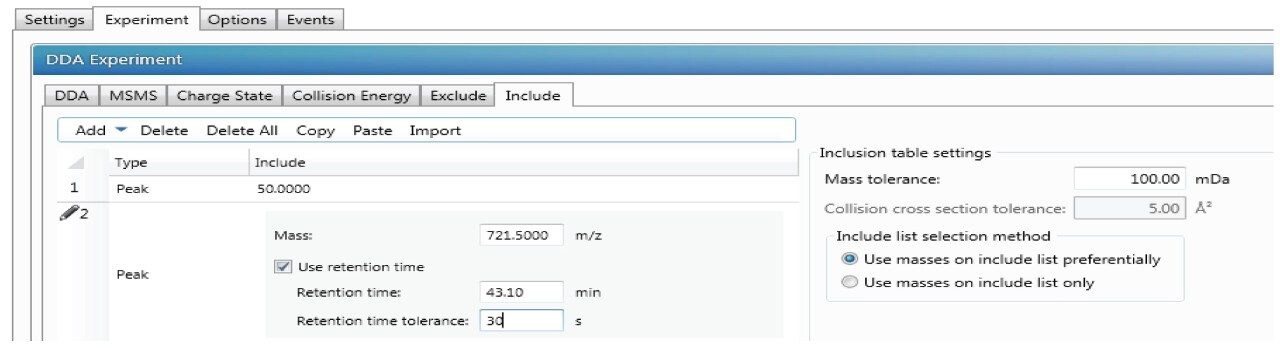
All unmodified Lys residues are susceptible to glycation in theory. However, the rate of individual modification varies depending on local sequence and higher order structure.2 The presence of glycation prevents trypsin and lys-C cleavage at the modified residue.3 Thus, up to 2 missed cleavages were considered during peak searching in UNIFI. In the DDA method, a lower intensity threshold of 5000 was used to trigger an MS/MS event. The low trigging threshold allows more low abundance precursor ions to be subjected to MS/MS fragmentation. In addition, a TIC threshold of 2e6 and a timeout of 30 sec were enabled to prevent the same high intensity precursor ions from undergoing repeated fragmentation. With these DDA parameters, more fragment ions were observed for low abundance ions such as the glycated peptides compared to MSE. The identified glycated peptides with modification levels from 0.05 to 4.77% cover 23 lysines in NIST mAbs. The identified glycated sites and the site occupancy are calculated by the MS response of glycated peptide divided by the sum of glycated peptide and unmodified peptide. An equation is shown below using a representative peptide. The results are listed and compared with the published result4 at Table 1.