Mass spectrometry-based quantification of therapeutic proteins uses a surrogate peptide as a stoechiometric representative of the protein which is cleaved. One of the challenges encountered in the quantification of therapeutic proteins in biological fluids (for example serum, plasma, and urine) is to find a suitable internal standard for each protein analyte. Isotopically labeled protein standards have been used successfully for quantification of mAbs,1-3 but these standards are expensive and time-consuming to produce.
Since protein quantification is performed at the peptide level, another alternative is to use relatively inexpensive isotopically labeled peptides as peptide IS.4-7 To account for trypsin digestion efficiency, 13C15N-isotopically labeled cleavable peptides (extended peptides) were introduced in 2004.5
Regardless of the nature of the IS, both quantification approaches rely on the digestion of the protein sample with a specific enzyme (such as trypsin, Lys C). Protein digestion has been recognized as the major source of variability in the analytical workflow4-9 which has to be carefully optimized. In addition, when the whole digest approach is implemented, using peptide IS for quantification, there is an additional concern related to the ability of the digestion enzyme to cleave with the same efficiency as the therapeutic protein as well as the isotopically labeled peptide IS, in the presence of the biological matrix.
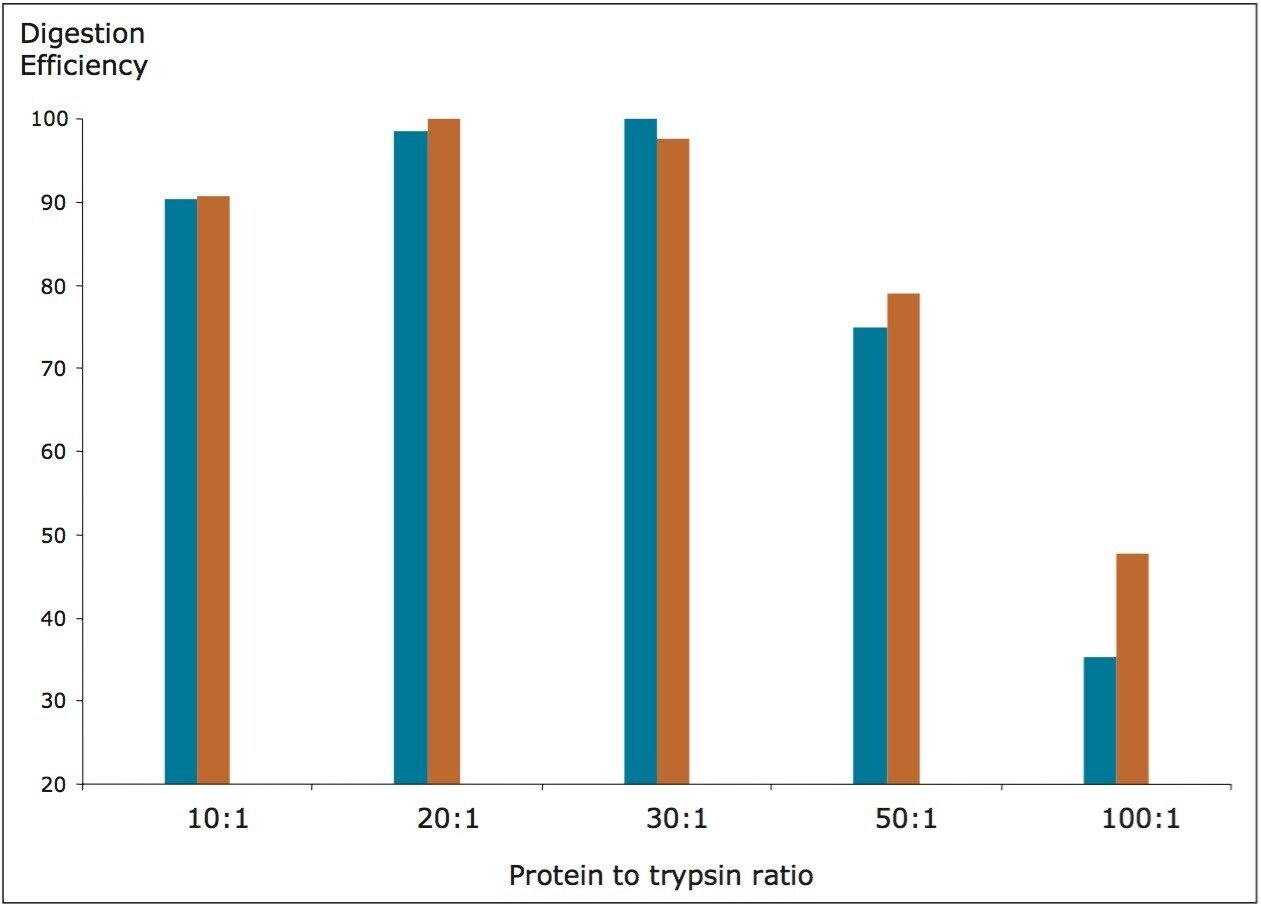
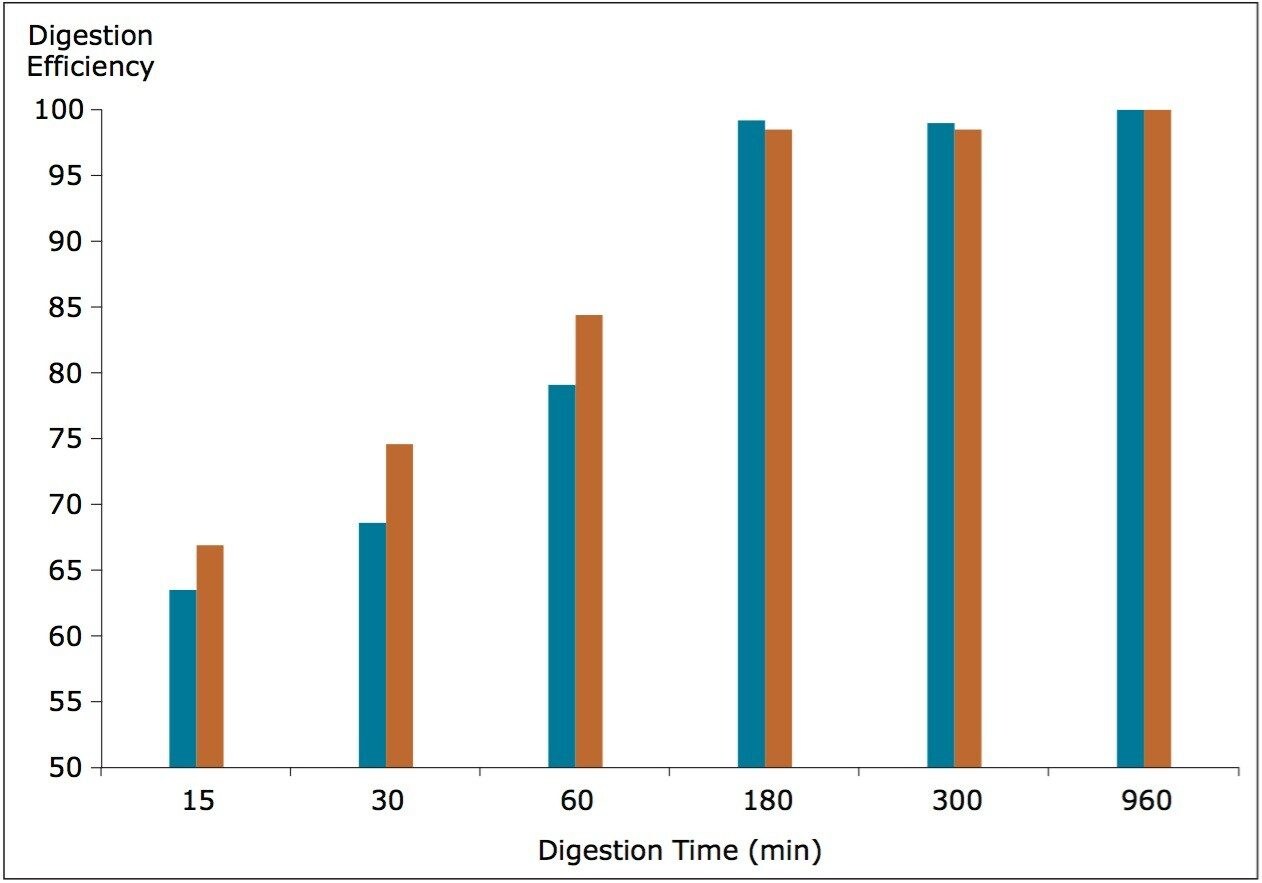
In this application, trypsin digestion optimization is required for developing a successful MRM assay. Generally, some of the benefits of trypsin digestion optimization include higher assay sensitivity, shorter sample preparation times, and higher assay reproducibility.
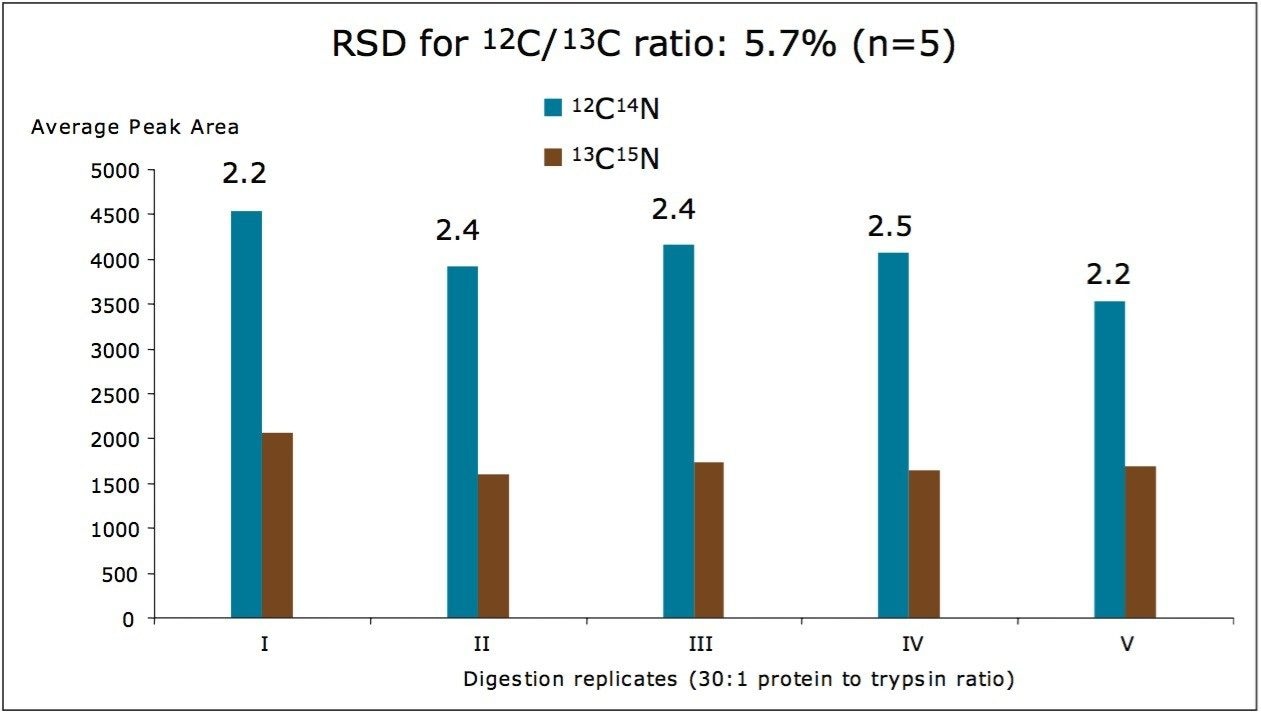
Here, we used a therapeutic mAb to investigate the trypsin digestion efficiency relative to the whole digest quantification method. Two digestion parameters, protein to trypsin ratio and digestion time, were optimized. Additionally, the reproducibility of the entire sample preparation protocol was assessed.
Trastuzumab (herceptin) is a humanized IgG1 kappa monoclonal antibody (mAb). The antibody was produced through genetic engineering10,11 by joining the constant regions of the human monoclonal antibody with the complementaritydetermining regions (CDRs) of a mouse monoclonal antibody able to bind human epidermal growth factor receptor 2 proteins (HER2) receptors. These HER2 receptors belong to a family of human oncoproteins expressed in approximately 25% of invasive breast cancers. Trastuzumab was approved in 1998 by the U.S. Food and Drug Administration (FDA) for the treatment of HER2-overexpressing breast cancers.
In this application note, we optimized the trypsin digestion step for the development of a sensitive LC-MRM assay for trastuzumab in human serum.