Macrolide antibiotics, such as spiramycin, tylosin, and erythromycin are basic, lipophilic molecules that consist of 14–16-membered lactone rings, to which sugar moieties are attached. Lincosamides, such as lincomycin and pirlimycin, are structurally very different from macrolides but share a similar mechanism of action. Macrolides and lincosamides are widely approved for use in veterinary medicine to treat respiratory diseases. Additionally, macrolides are licensed for use in some countries and regions as feed additives to increase the conversion rate of feed and promote animal growth.
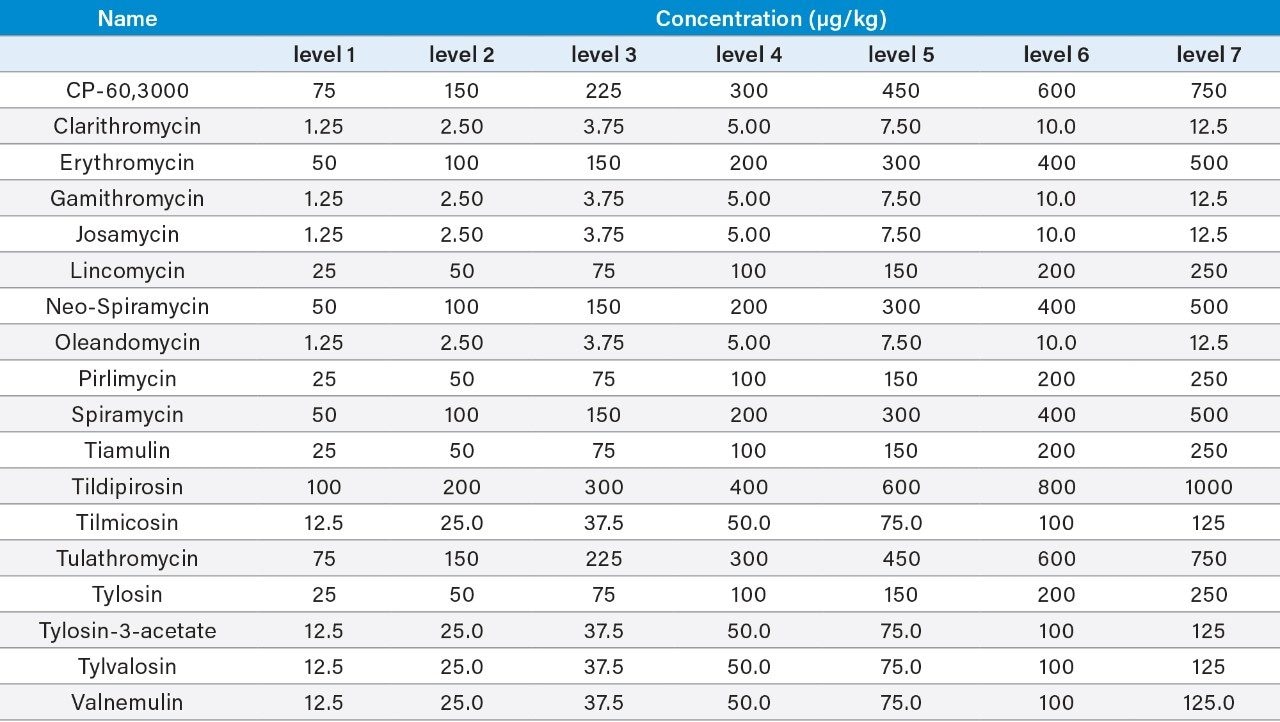
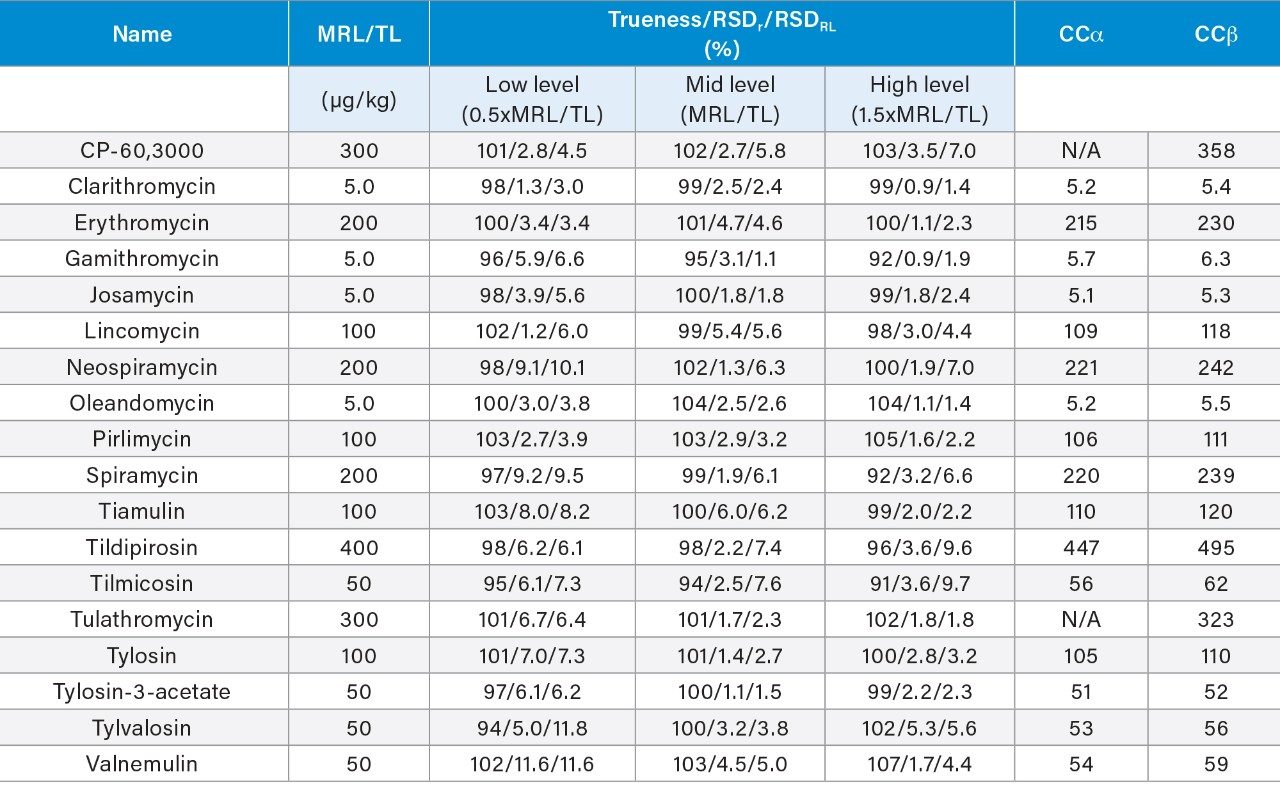
Although veterinary drugs play an important role in the production of livestock and poultry, incorrect use of macrolides or the shortening of the withdrawal time after treatment can possibly lead to the presence of macrolide residues in animal tissues and related foodstuffs and increases the potential risk to consumers because of allergic reactions of those sensitive to the antibiotics. To protect human health, rules exist to ensure consumer protection against the potentially harmful effects of residues in foodstuffs of animal origin (e.g., Regulation (EU) 2019/6 in the EU).1 Legislation provides for a science-based establishment of maximum residue limits (MRLs) for veterinary medicinal products. A maximum residue limit is the maximum concentration of a residue of a pharmacologically active substance that may be permitted in food of animal origin. MRLs for macrolides vary widely depending on the animal and target tissue and the country setting them.2,3 As macrolides are produced from various Streptomyces strains, they tend to be multi-component systems containing lower amounts of related compounds. For example, tylosin consists predominantly of tylosin A but with varying amounts of desmycosin (tylosin B), macrocin (tylosin C), and relomycin (tylosin D). The marker residue tends to be the most abundant component found in the tissue (e.g., tylosin A for tylosin). Since tulathromycin and most of its metabolites can be converted by acid hydrolysis to a metabolite known as CP-60,300, the EU chose this compound as the marker residue for tulathromycin, and established MRLs defined as the sum of tulathromycin and its metabolites that are converted by hydrolysis to the marker residue (CP-60,300) and expressed in tulathromycin equivalents. The inclusion of a hydrolysis step is not feasible when including multiple macrolides in one method, so here we consider this method suitable for screening tulathromycin.
In the case of a suspected non-compliant result, analysis is repeated using a validated confirmatory method, with a hydrolysis step, that complies with the residue definition. Residue monitoring plans are used to detect the illegal use or misuse of authorized veterinary medicines in food-producing animals and investigate the reasons for residue violations. In some cases, such as in the EU, exporting countries must also implement a residue monitoring plan that guarantees an equivalent level of food safety.
Food business operators also undertake chemical analyses to check for the presence of residues in tissues of animals within their supply chain for due diligence and positive release purposes. In addition to checking MRL compliance, there is growing concern about antibiotic resistance and its threat to human health. Acquired resistance to macrolides and lincosamides among food animal pathogens, including some zoonotic bacteria, has now emerged.4
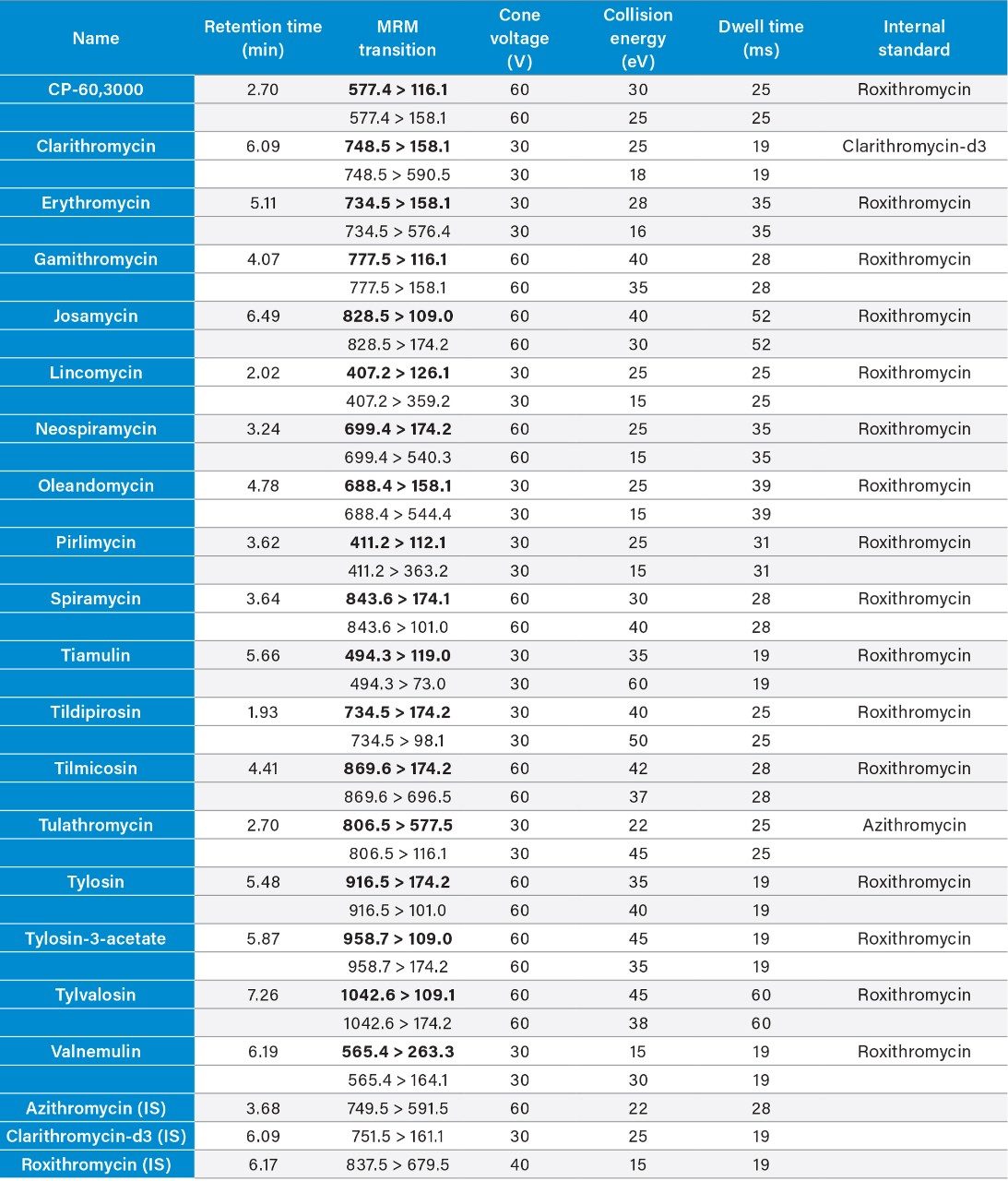
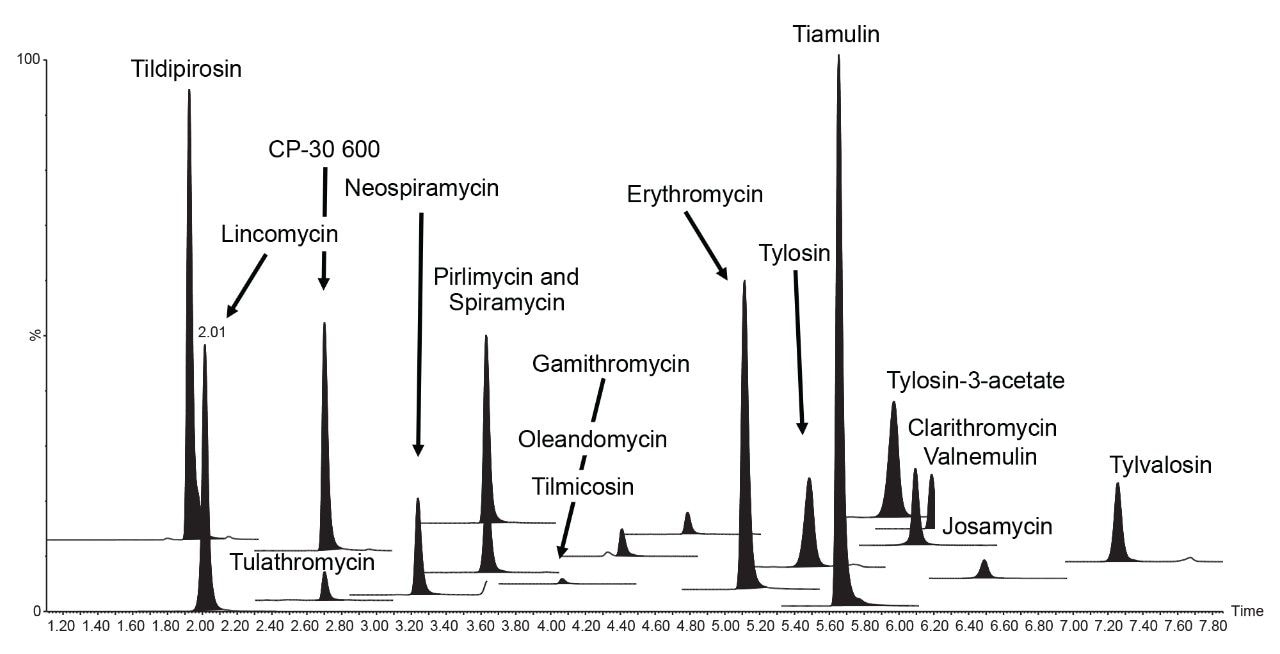
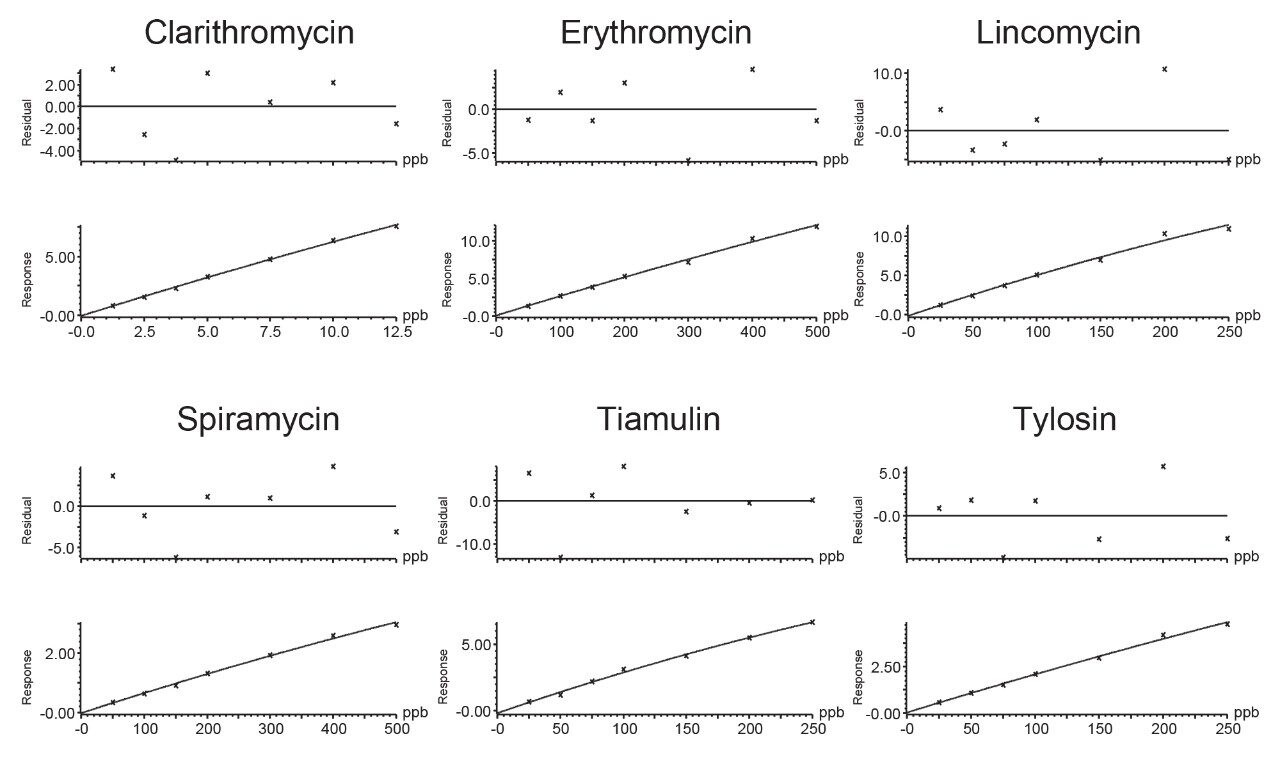
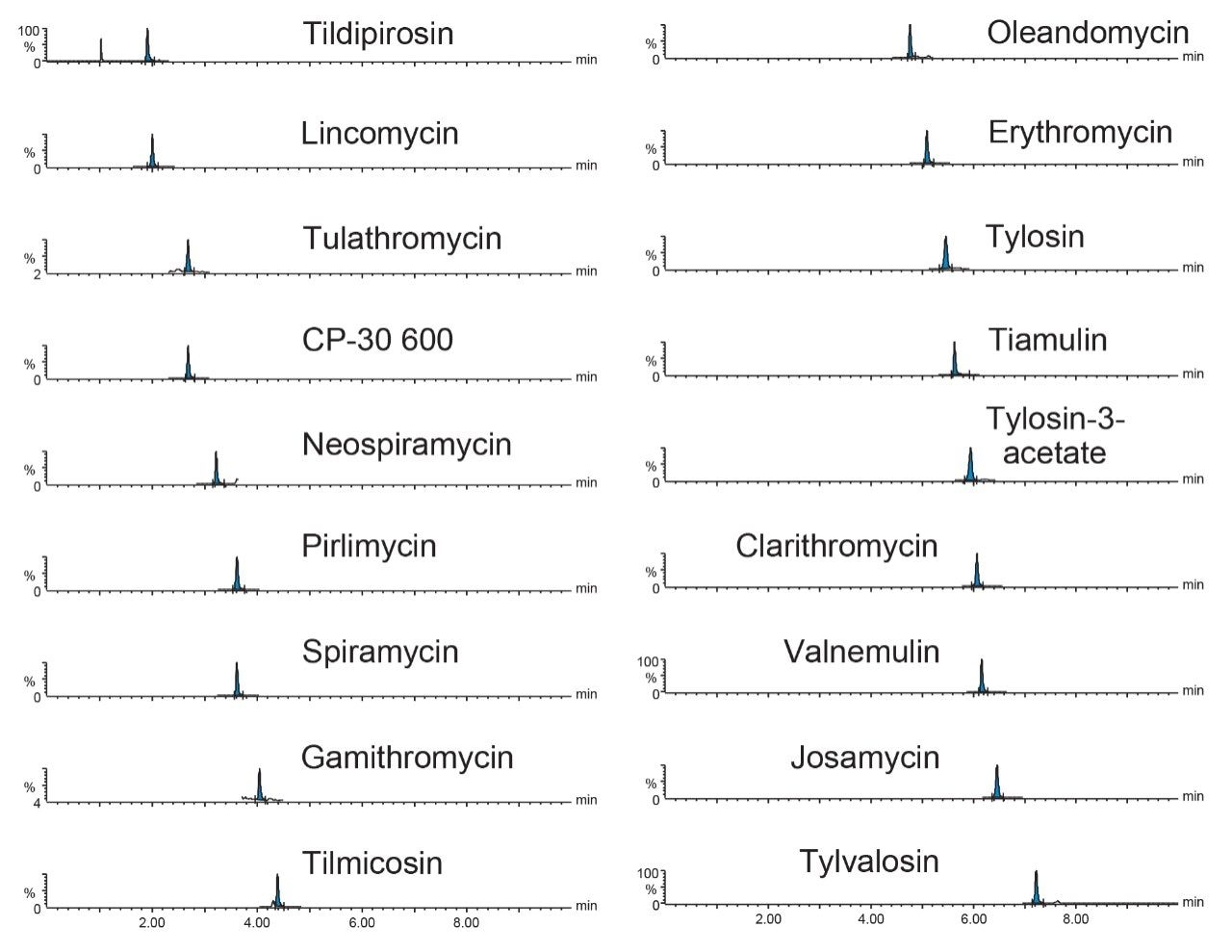
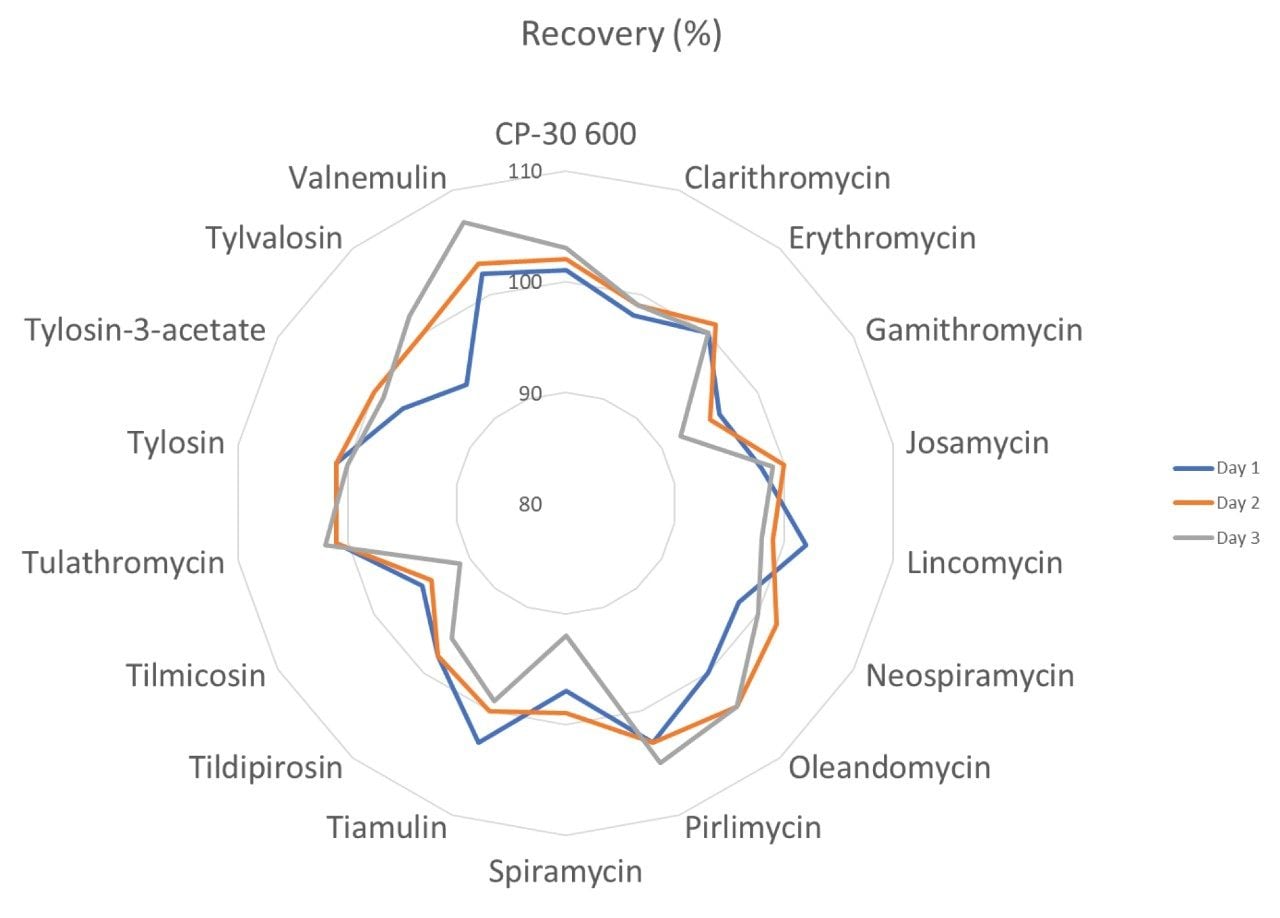
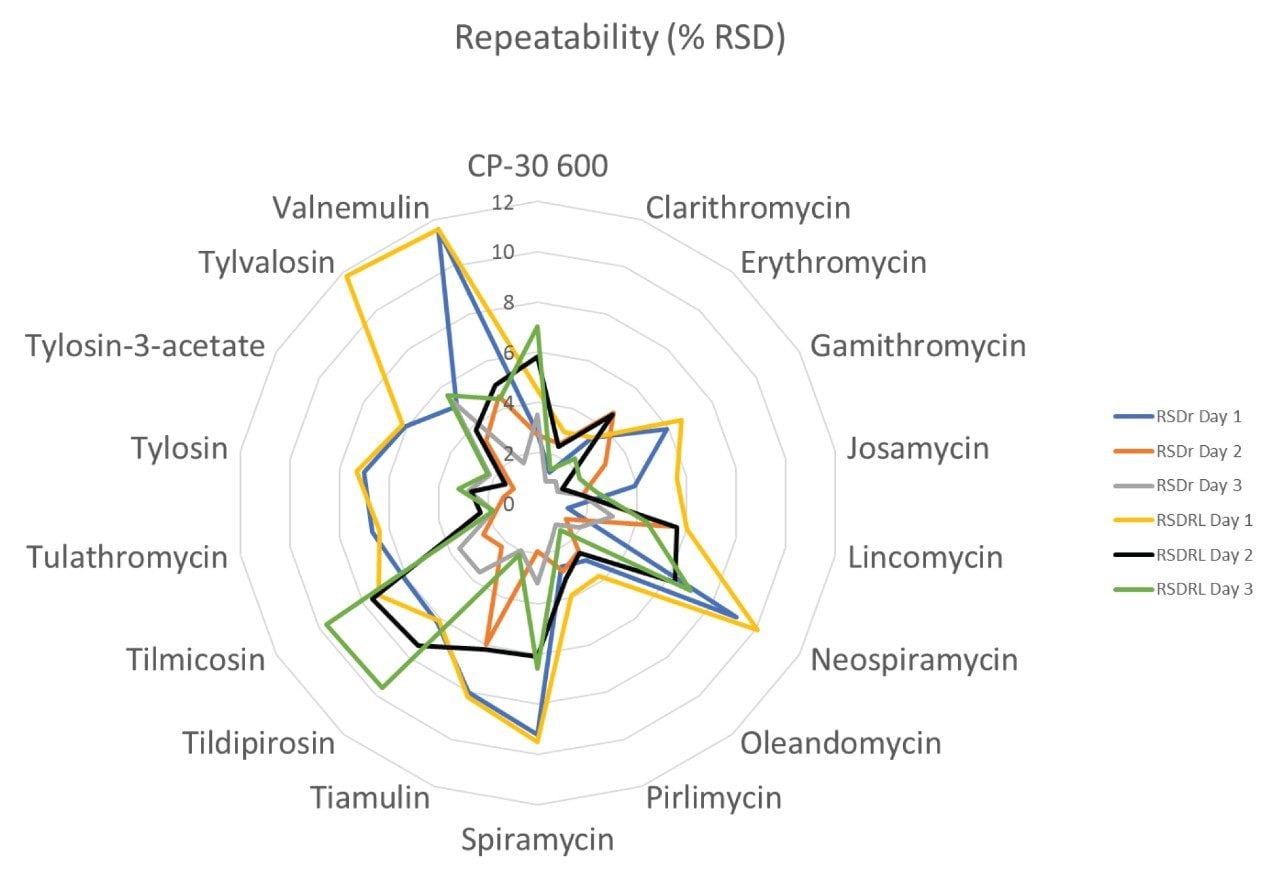
Therefore, it is important to develop simple but accurate methods for the determination of residues of antibiotics in animal tissues. This application note describes the validation of a method for the determination of 18 macrolide antibiotic veterinary drugs in bovine muscle tissue using the Waters ACQUITY UPLC I-Class PLUS coupled to the Xevo TQ-S micro.