Sample preparation
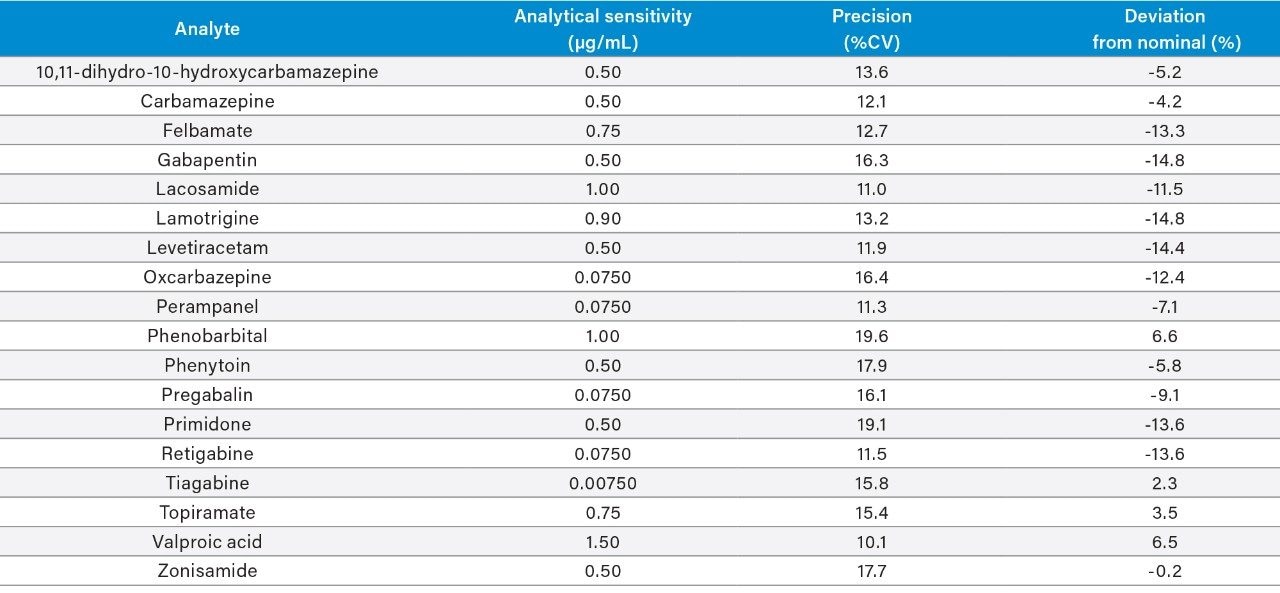
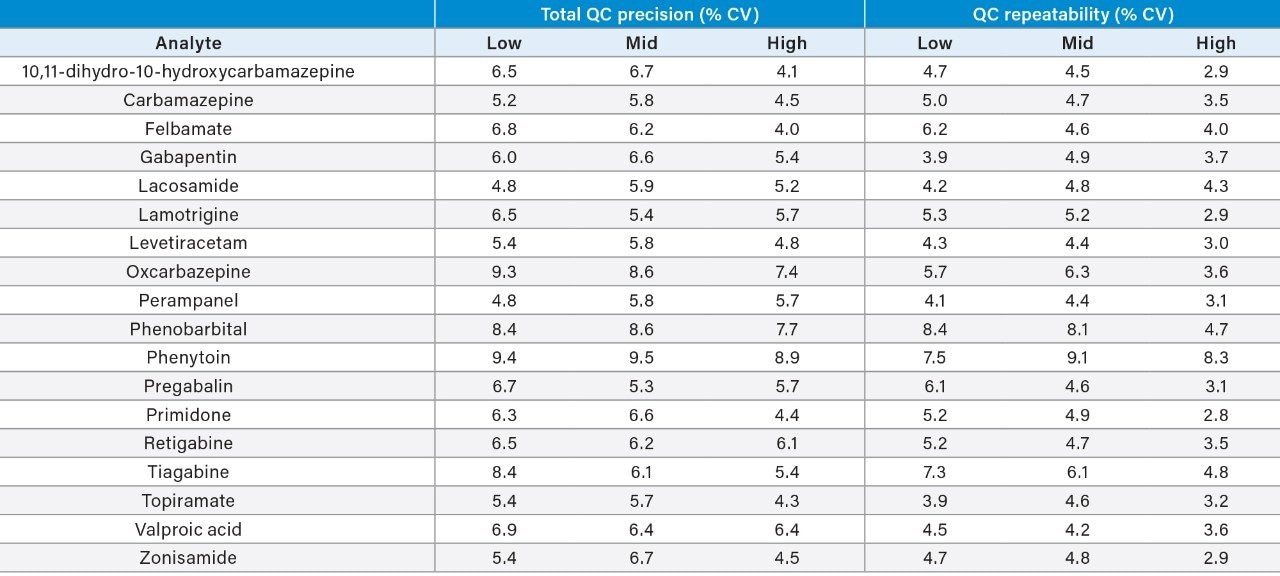
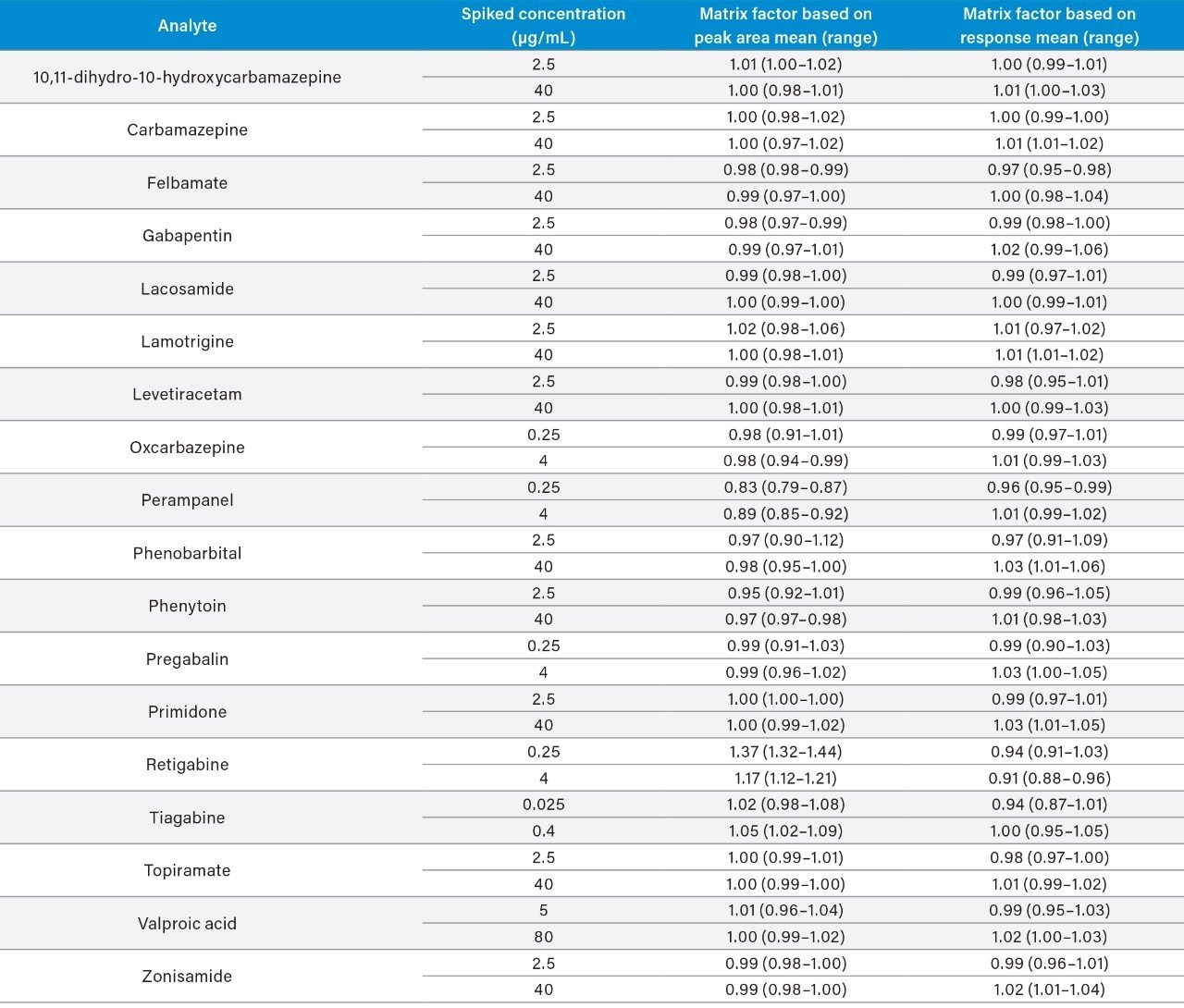
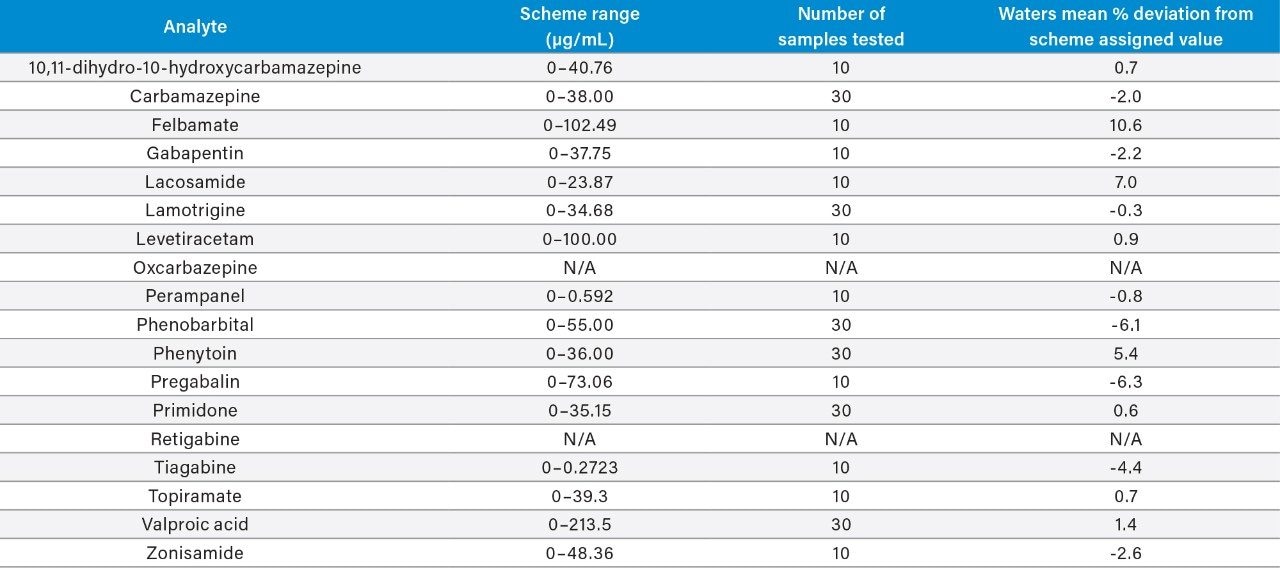
Plasma calibrators and quality control materials were prepared in-house using pooled human plasma supplied by BioIVT (West Sussex, UK). Concentrated stock solutions were prepared from certified powders and solutions supplied by Cambridge Bioscience (Cambridgeshire, UK), Fisher (Loughborough, UK), Sigma-Aldrich (Dorset, UK), and Toronto Research Chemicals (Ontario, Canada). Stable labelled internal standards were supplied by Cambridge Bioscience (Cambridgeshire, UK), Sigma-Aldrich (Dorset, UK), and Toronto Research Chemicals (Ontario, Canada). The calibration range was 1–100 μg/mL for all analytes, except for oxcarbazepine, perampanel, and pregabalin (0.1–10 μg/mL), tiagabine (0.01–1 μg/mL), and valproic acid (2–200 μg/mL). In-house quality control samples were prepared in plasma at low, medium, and high concentrations of 2.5, 7.5, and 40 μg/mL for 10,11-dihydro-10-hydroxycarbamazepine, carbamazepine, felbamate, gabapentin, lacosamide, lamotrigine, levetiracetam, phenobarbital, phenytoin, primidone, topiramate, and zonisamide; 0.25, 0.75, and 4 μg/mL for oxcarbazepine, perampanel, pregabalin, and retigabine; 0.025, 0.075, and 0.4 μg/mL for tiagabine; and 5, 15, and 80 μg/mL for valproic acid.