Peptides have many attractive attributes as druggable compounds, but frequently suffer from poor pharmacological properties, such as permeability and stability, which make their development challenging. To overcome these limitations, peptides are being increasingly engineered or enhanced through the introduction of more complex structures and non-native modifications.
Identifying catabolites of these peptides is an important function of the drug development process, in order to understand clearance and metabolite fate, particularly in the context of unique human catabolites. Confident identification is analytically and computationally challenging; even simple sequences contain many hydrolyzable bonds that lead to hundreds of potential catabolites. For in vitro or in vivo studies, various amino acids or engineered peptide segments may also face additional transformative pathways such as oxidations, deamidations, conjugation and deconjuction. The multiply charged nature of peptides acquired using an electrospray based LC-MS analysis leads to a large number of m/z values, all of which need to be screened against to ensure that all catabolites are identified. All of these factors add complexity to peptide catabolite identification.
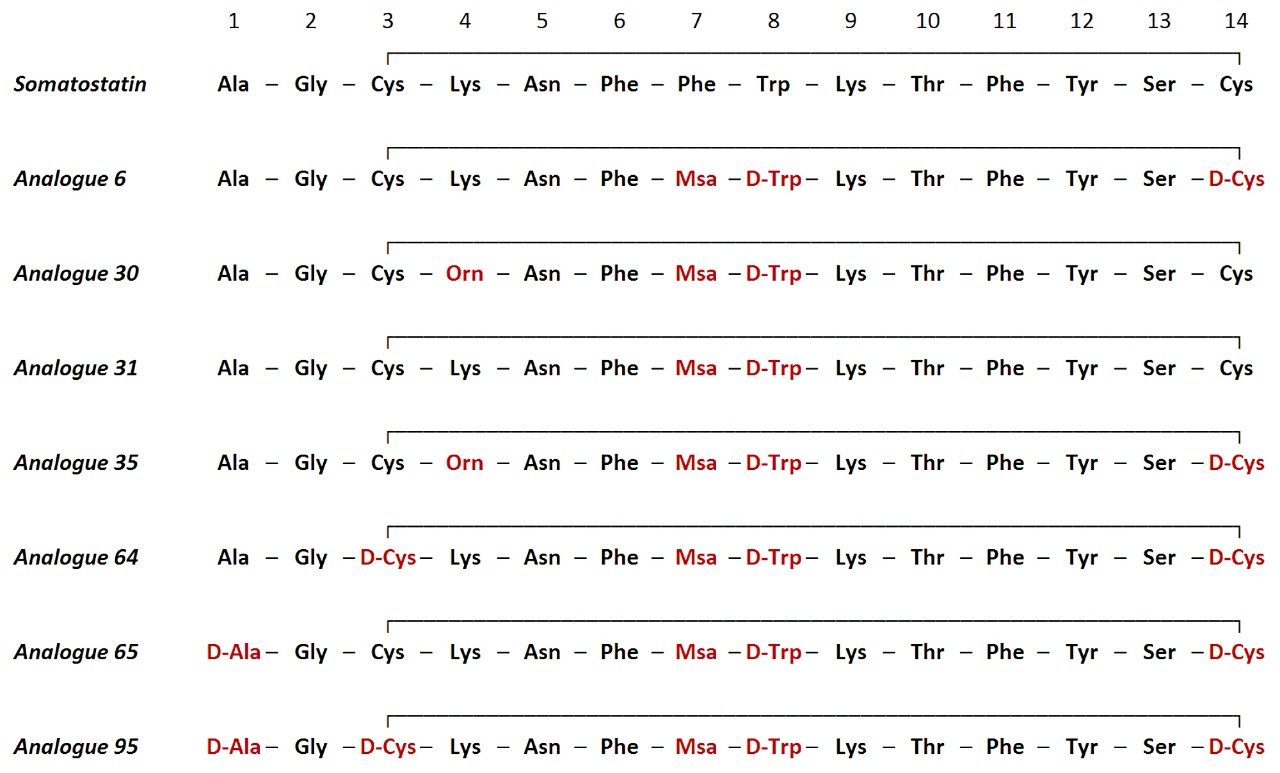
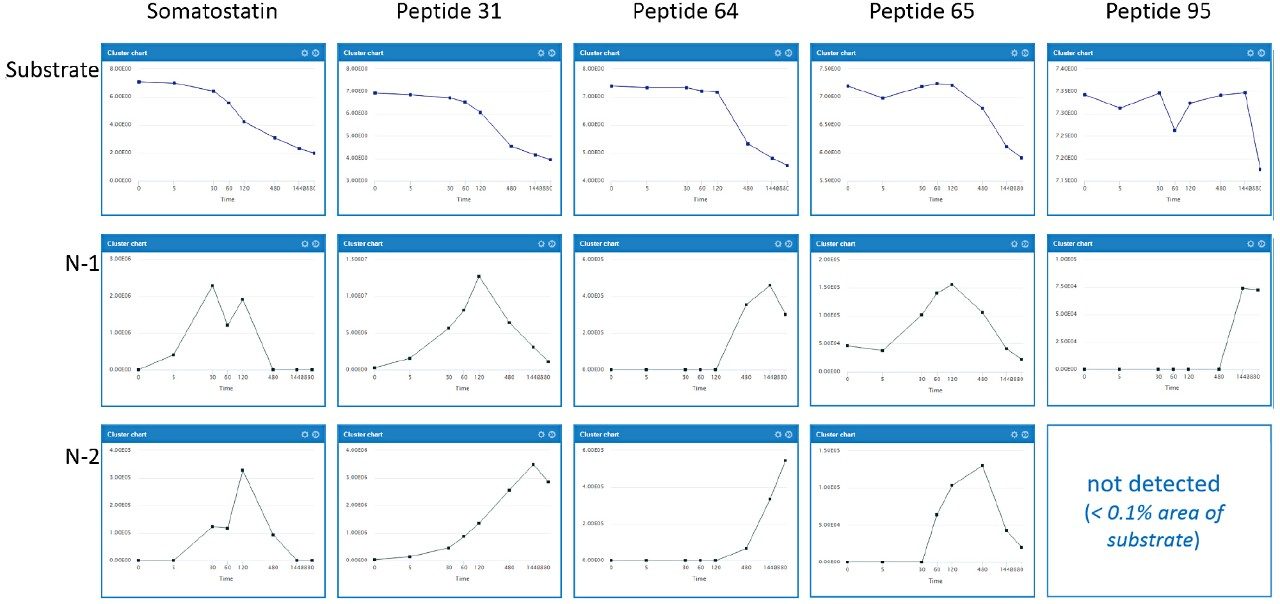
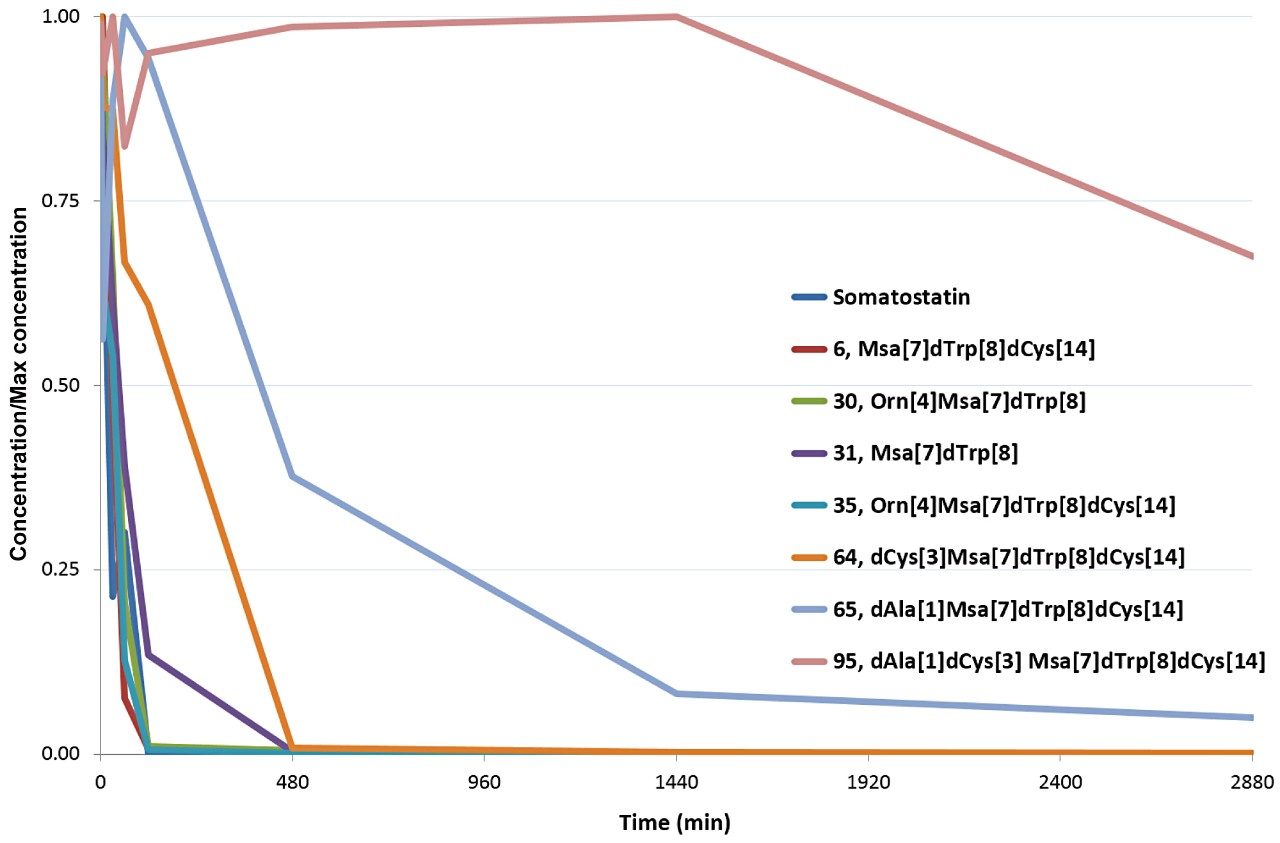
Somatostatin is a natural growth-inhibiting peptide hormone that has been the focus of numerous studies. Riera et al., published detailed descriptions of somatostatin and the influence of several sequence modifications on stability in human serum.3 In this application note, several of these serum samples were reanalyzed using ion mobility enabled high resolution mass spectrometry.
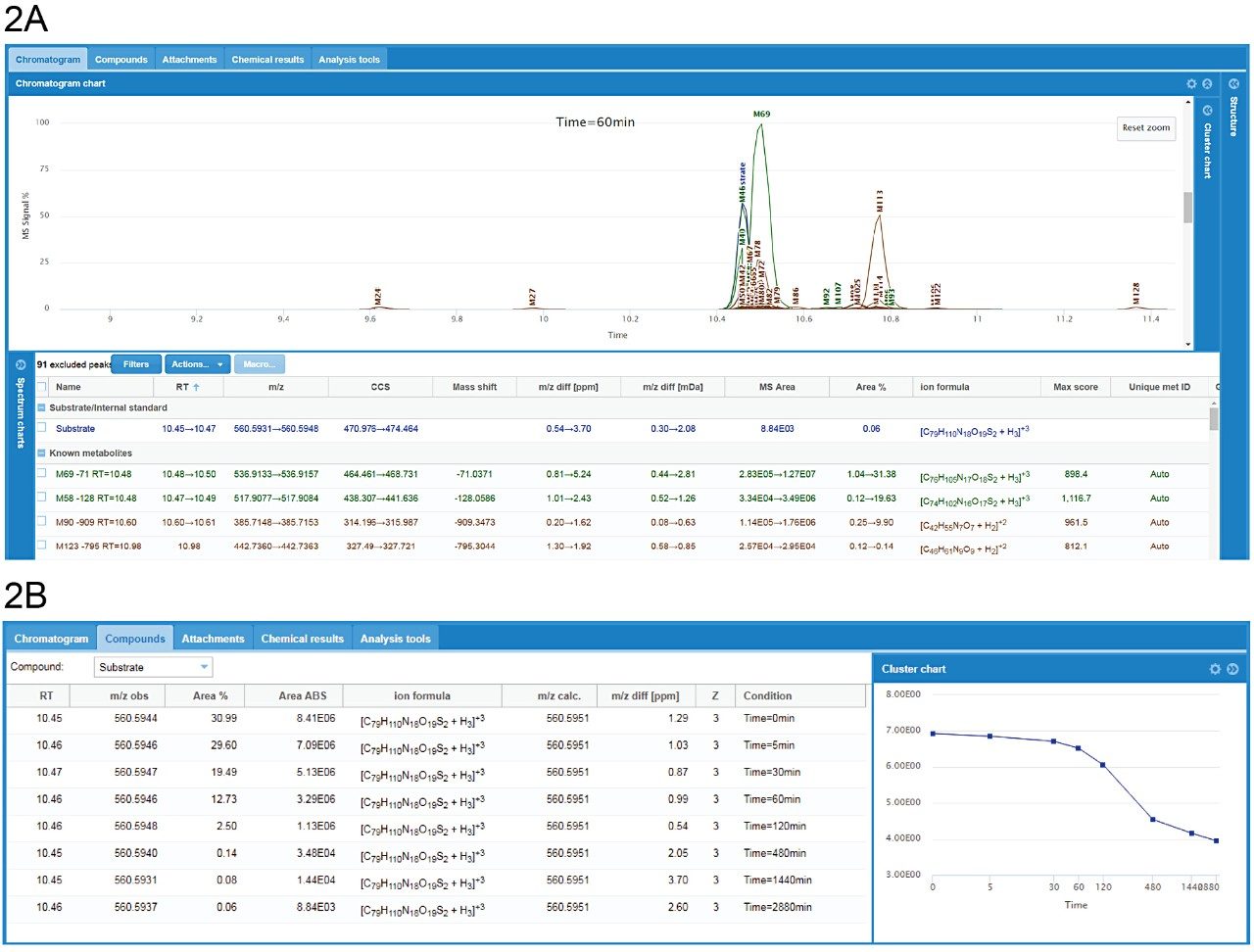
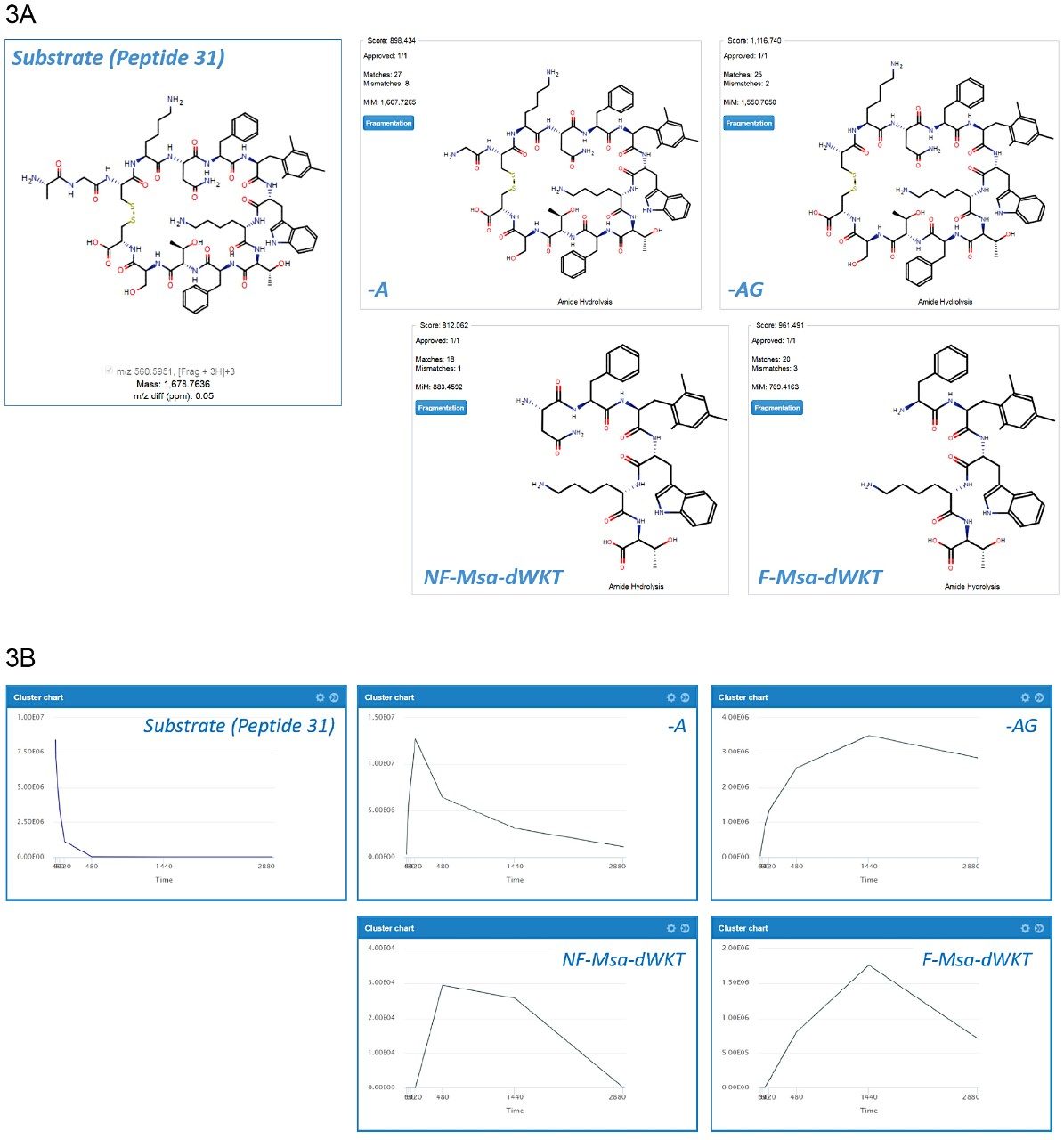
Data were processed using the software packages Mass-MetaSite and WebMetabase (Molecular Discovery Ltd.) to automatically detect and visualize peptide catabolites. When combined with the accuracy and selectivity of HDMSE acquired data (ion mobility enhanced HRMS data independent acquisition), this allows the confident identification and tracking of catabolites.4
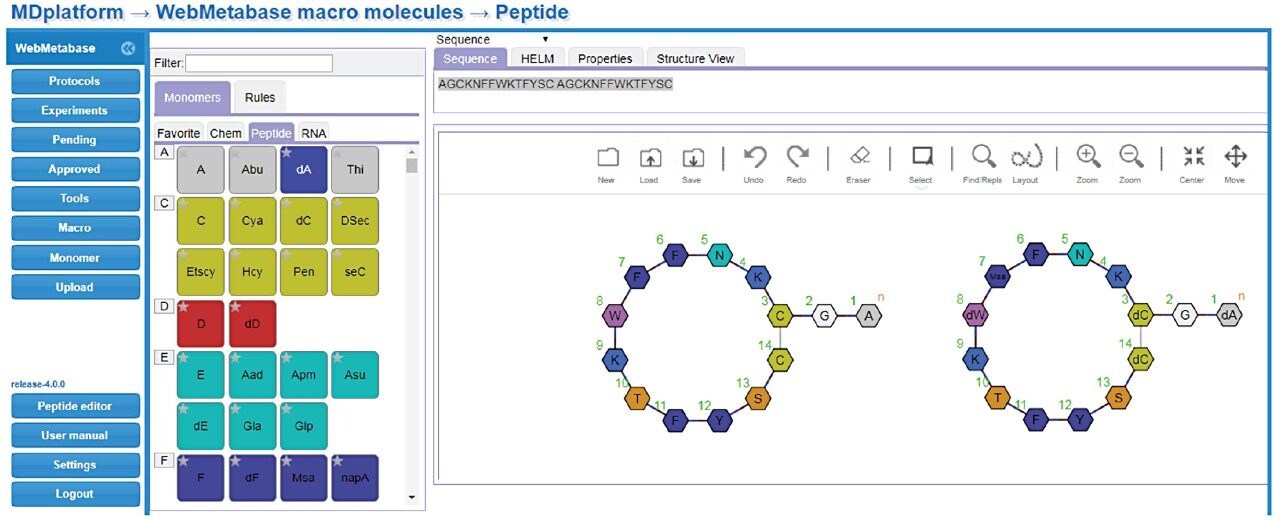
Studies were analyzed using a centralized server running WebMetabase based in the UK, enabling multiple global users and labs in both the UK and USA to upload, process, view data simultaneously, and serve as a single repository for datasets. Additionally, functionality in Webmetabase enabling HELM notation (Pistoia Alliance) proved useful for visualizing complex peptide catabolite and fragment identification.