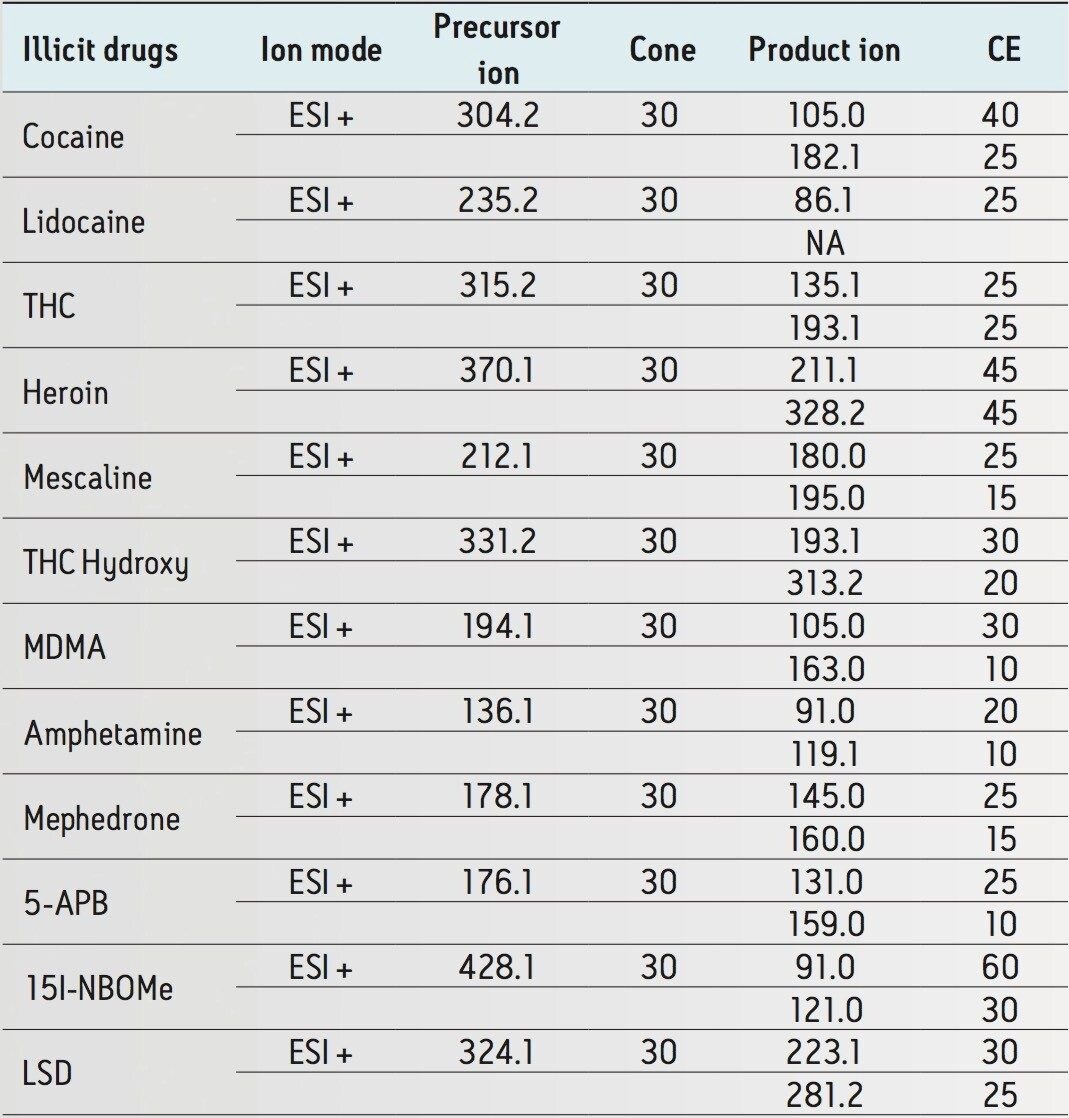
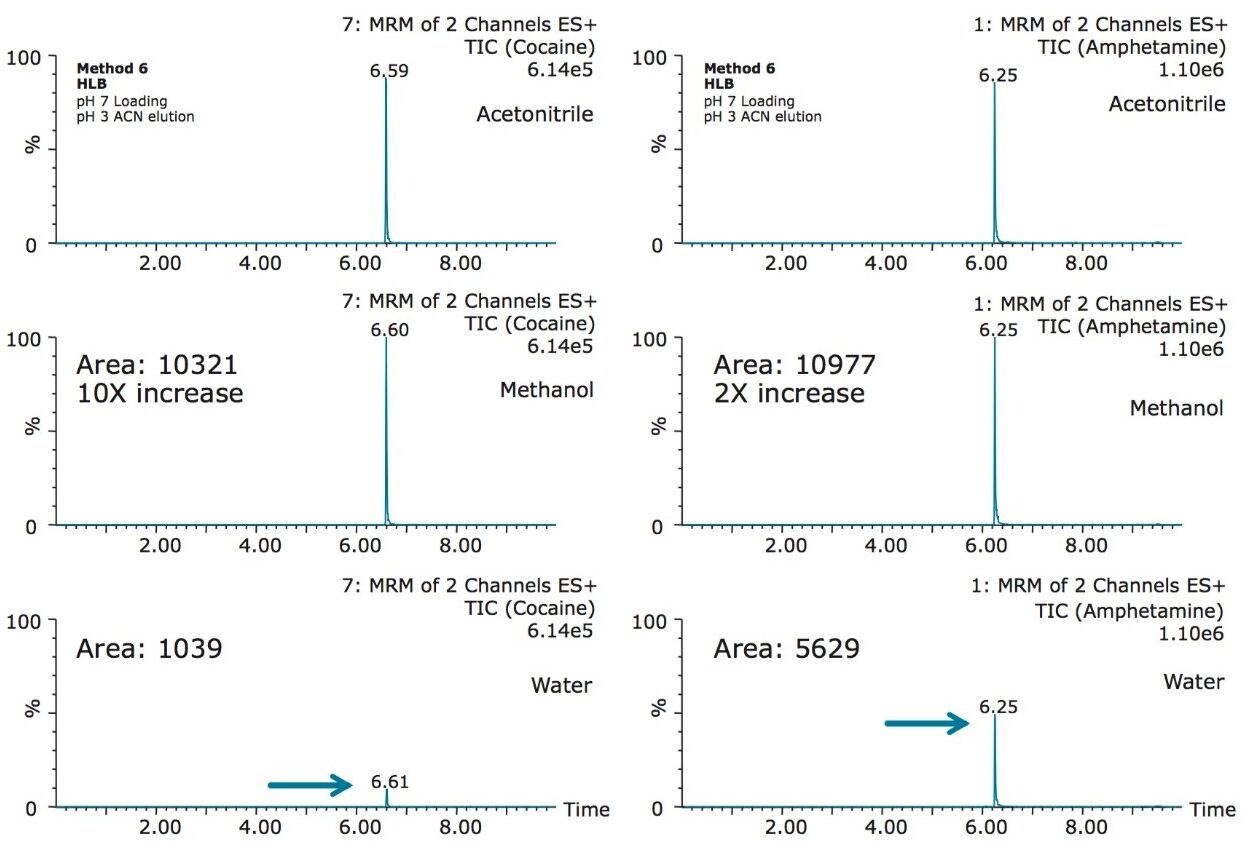
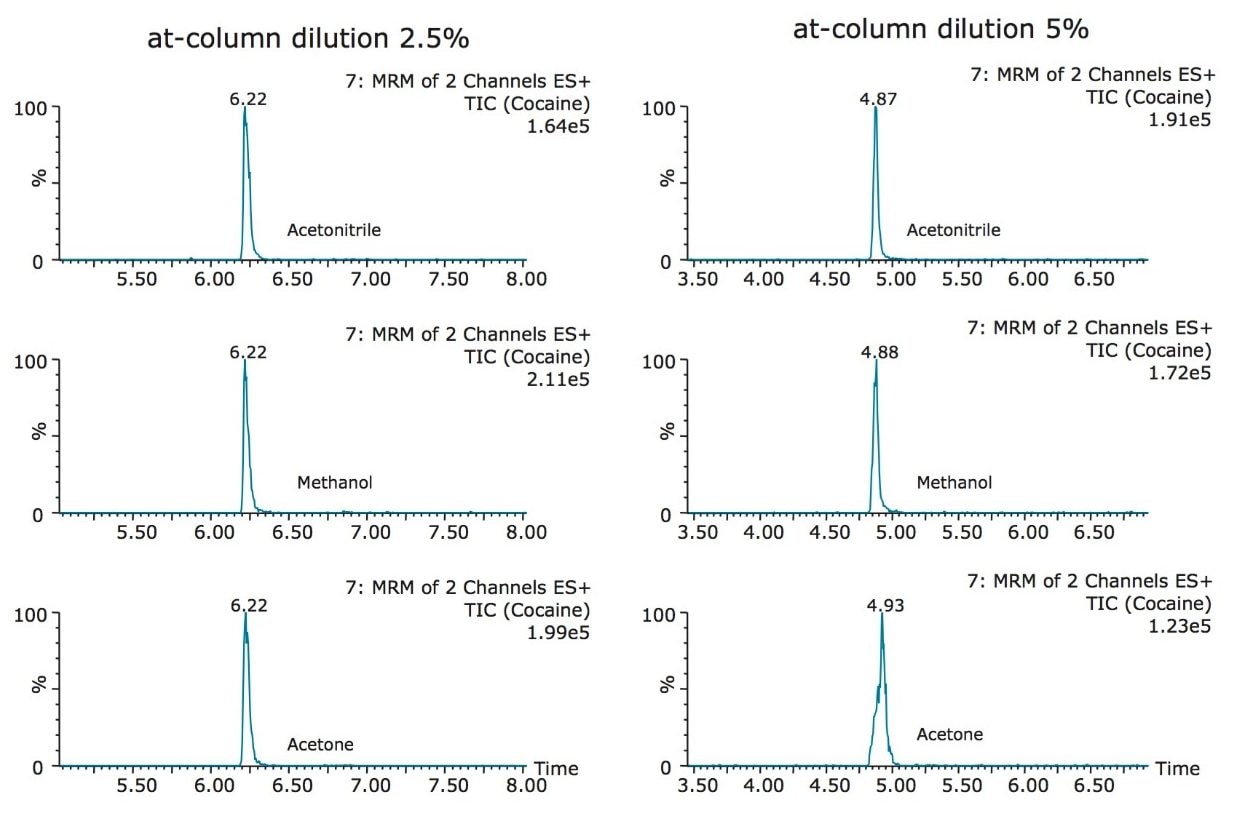
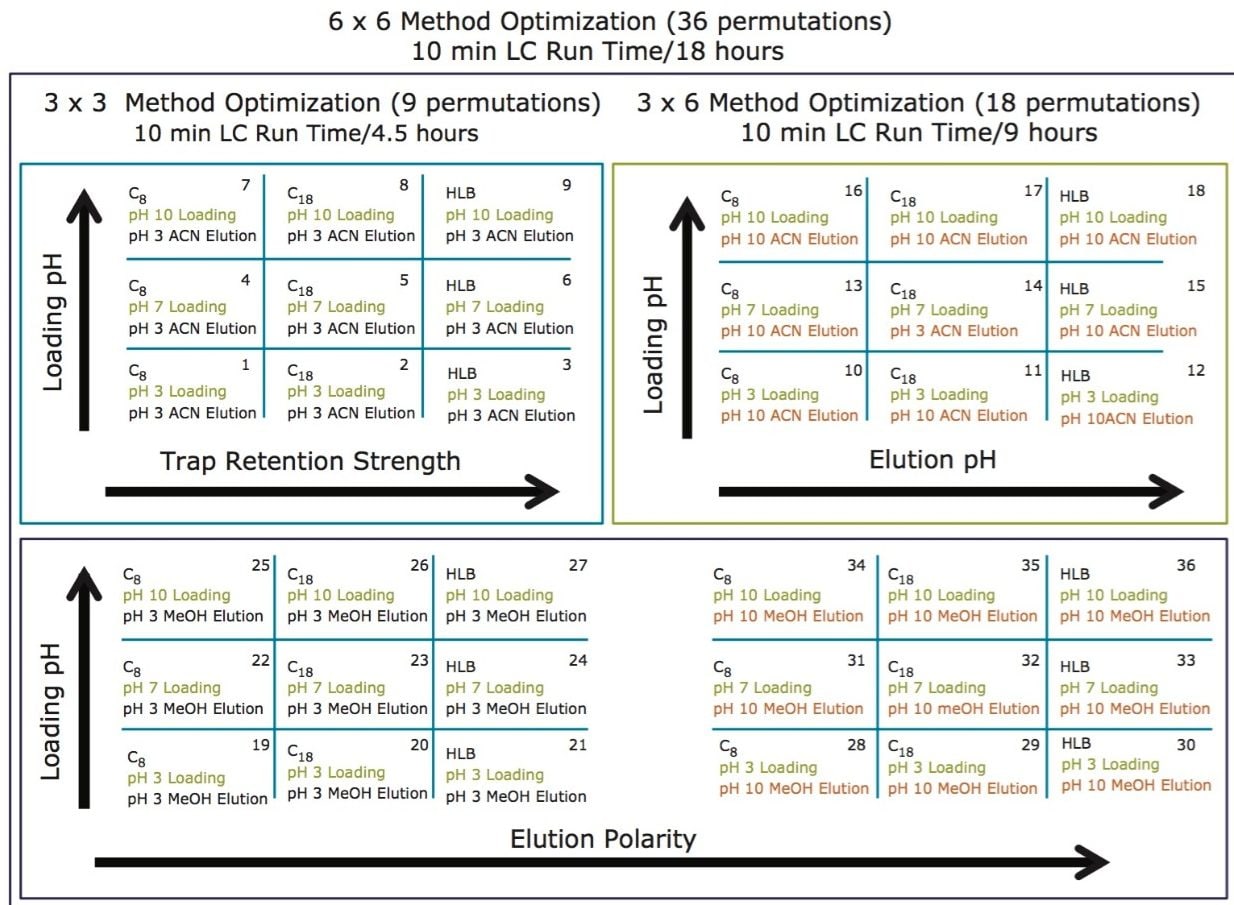
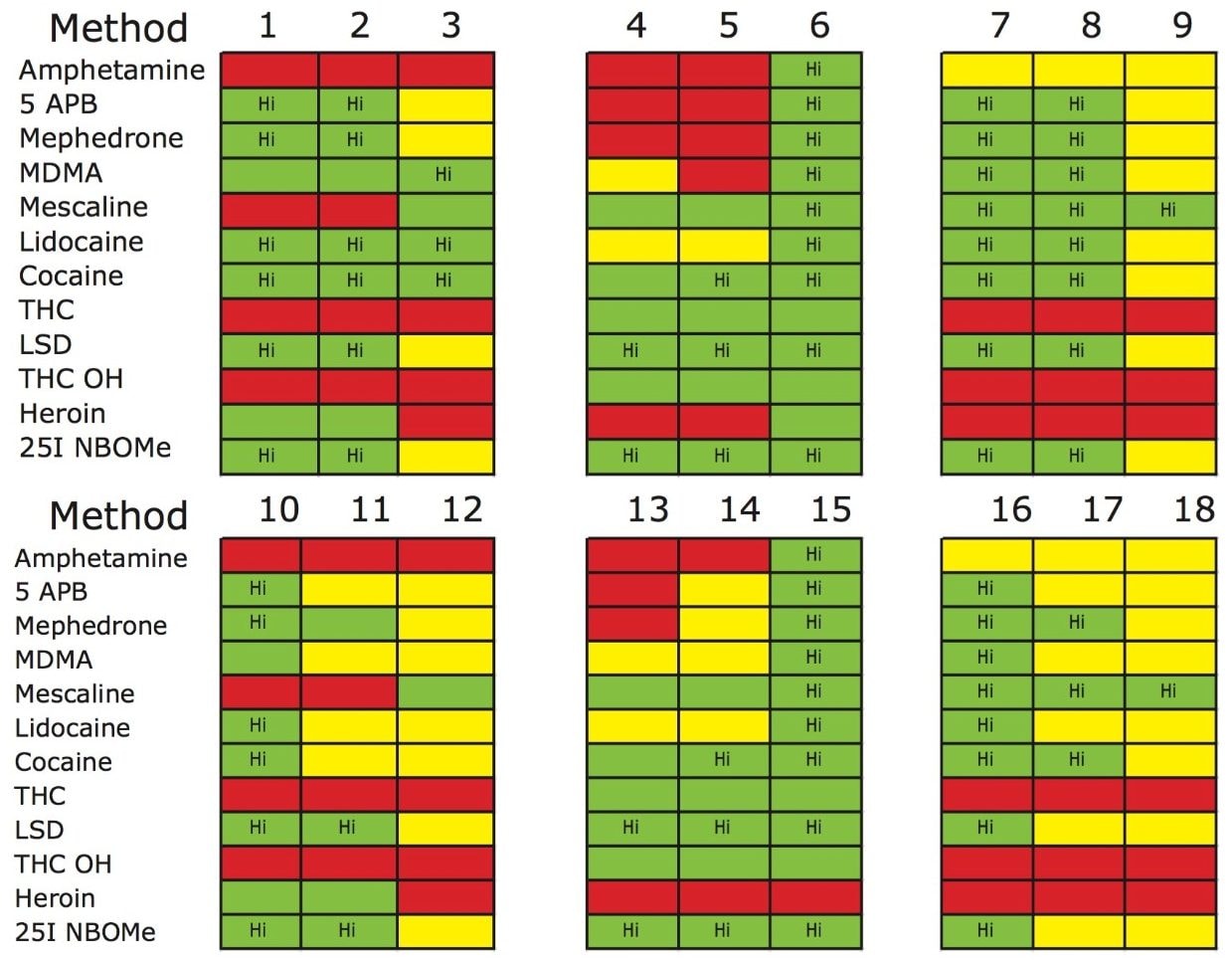
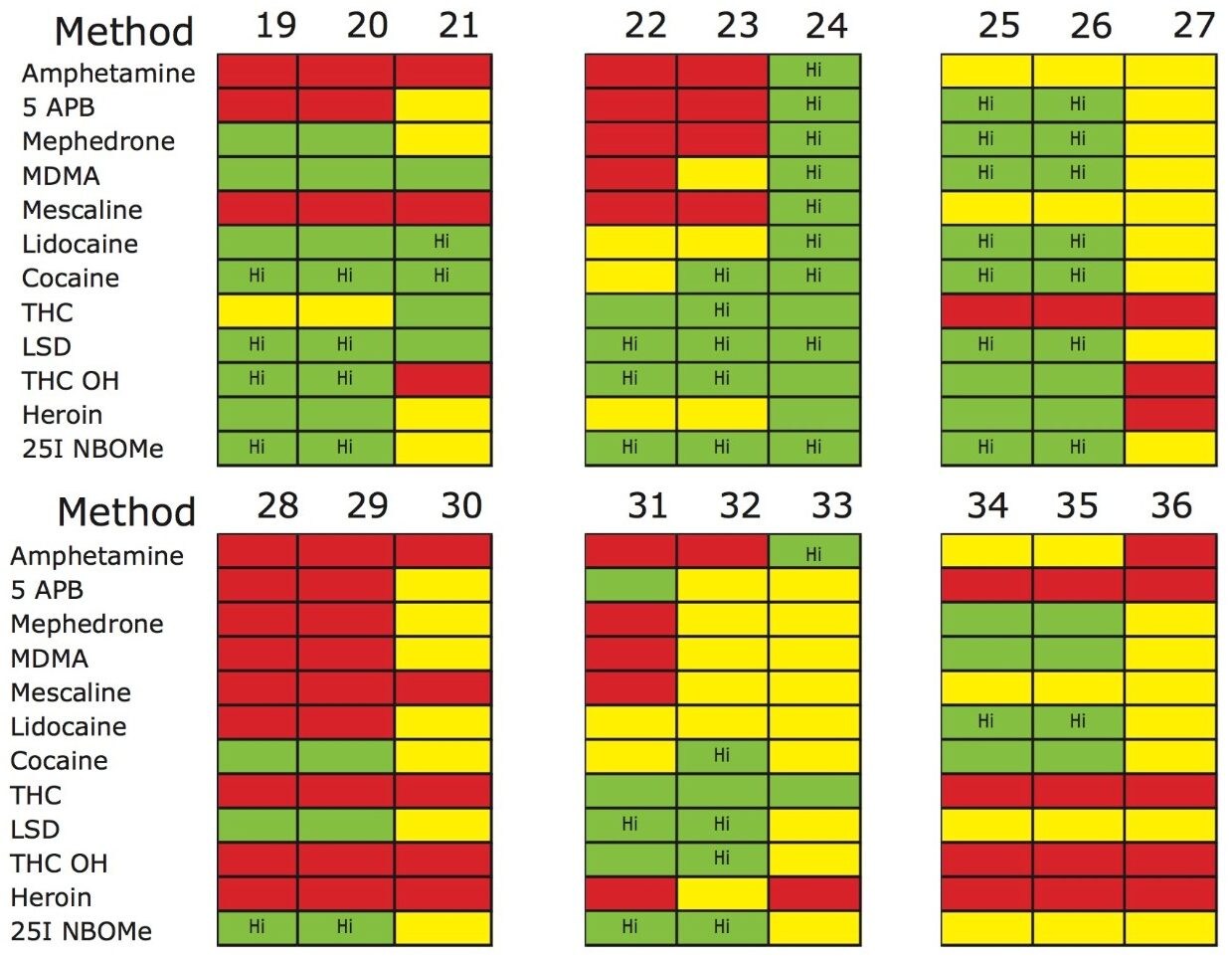
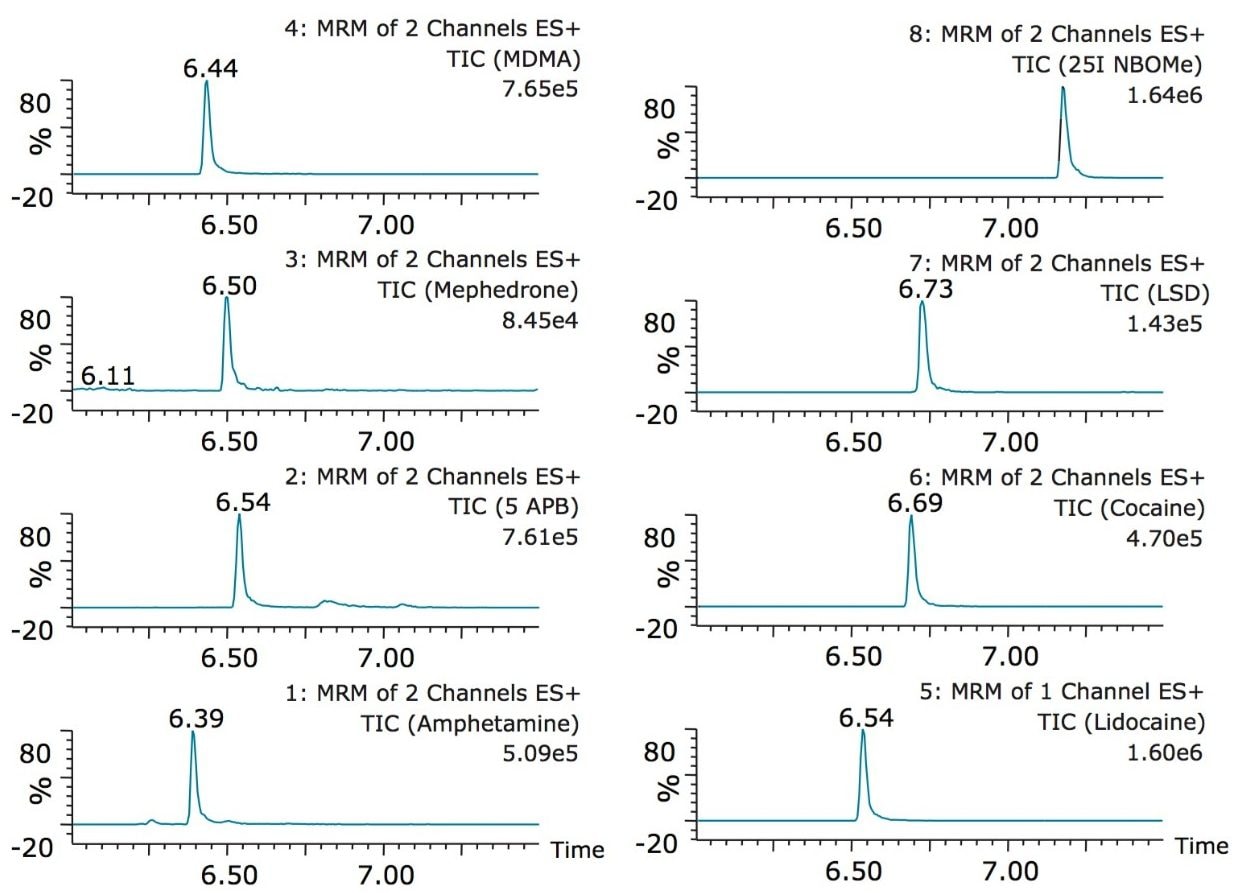
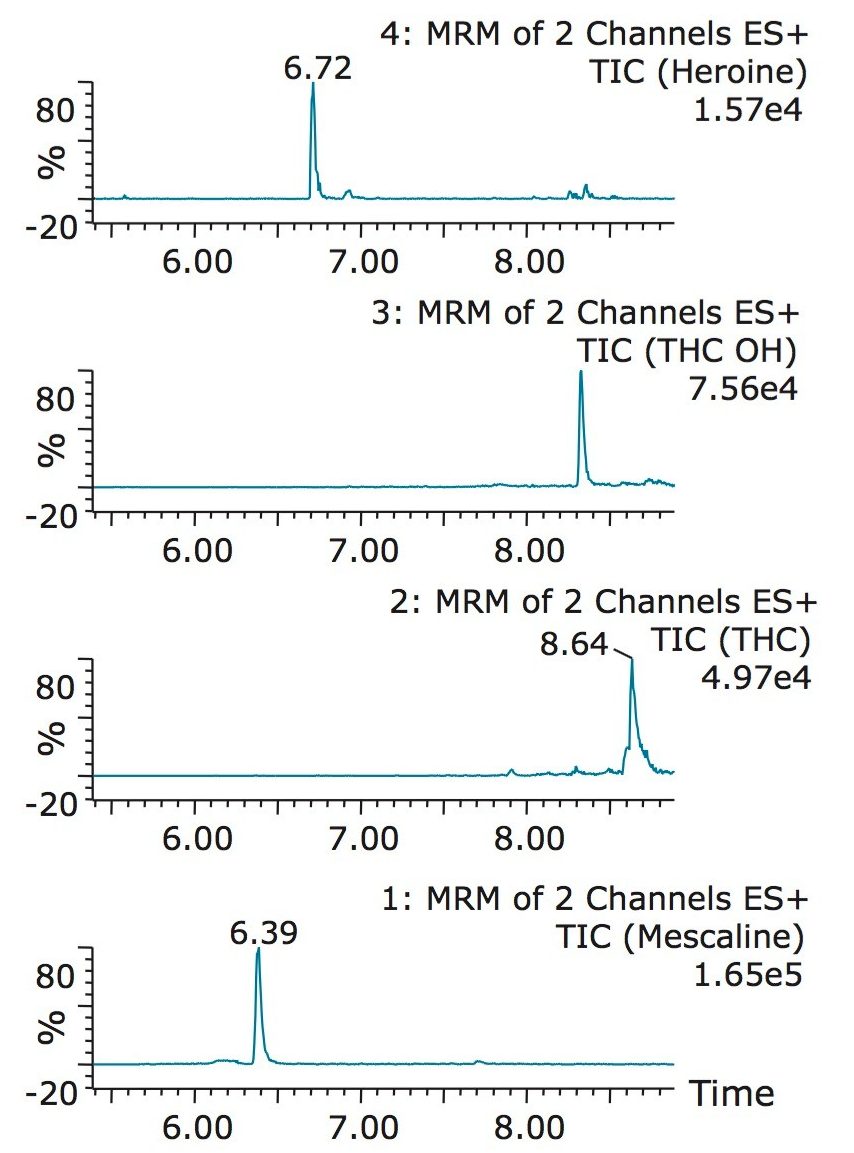
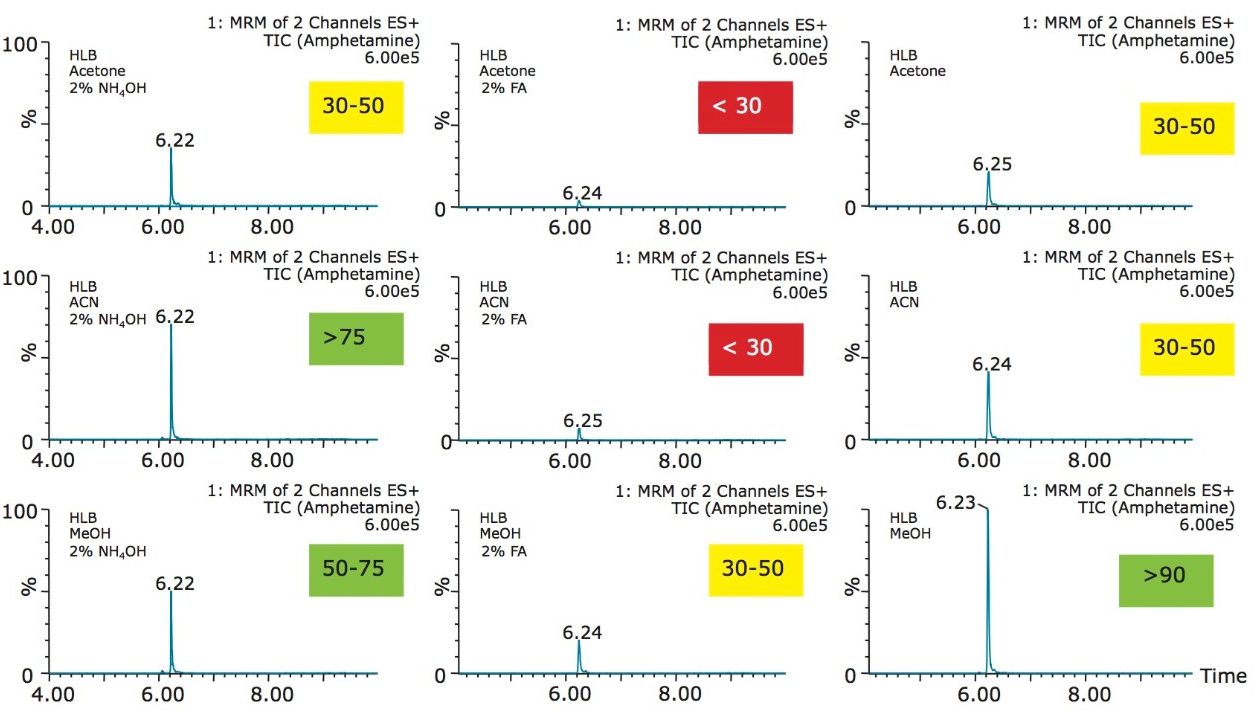
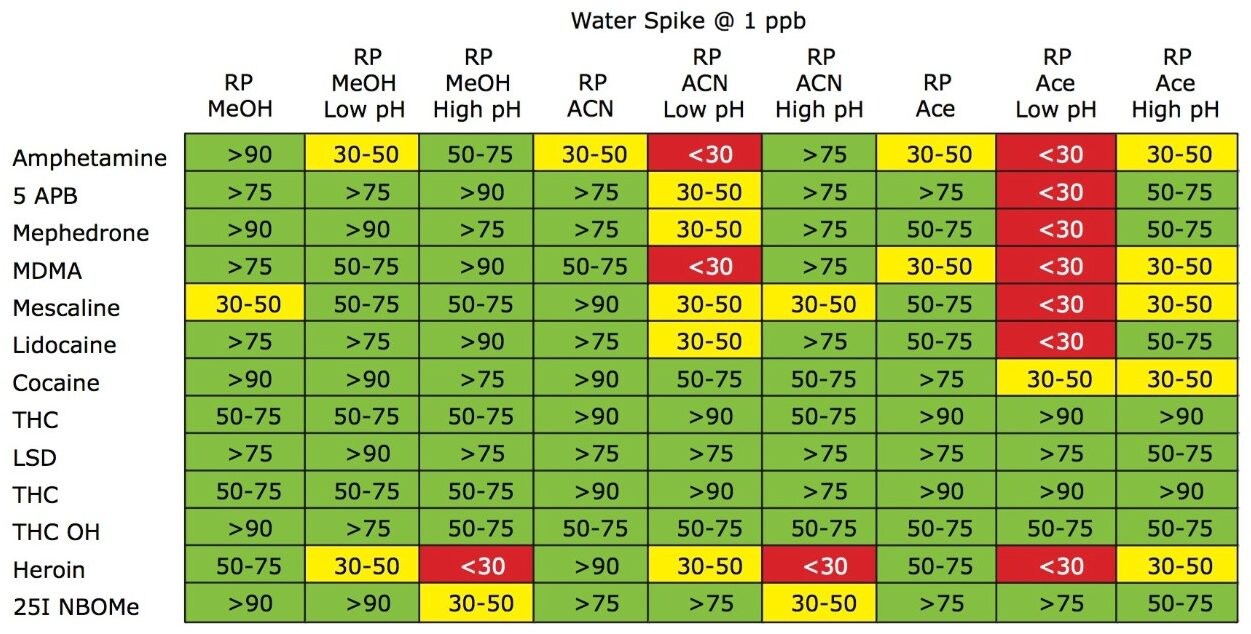
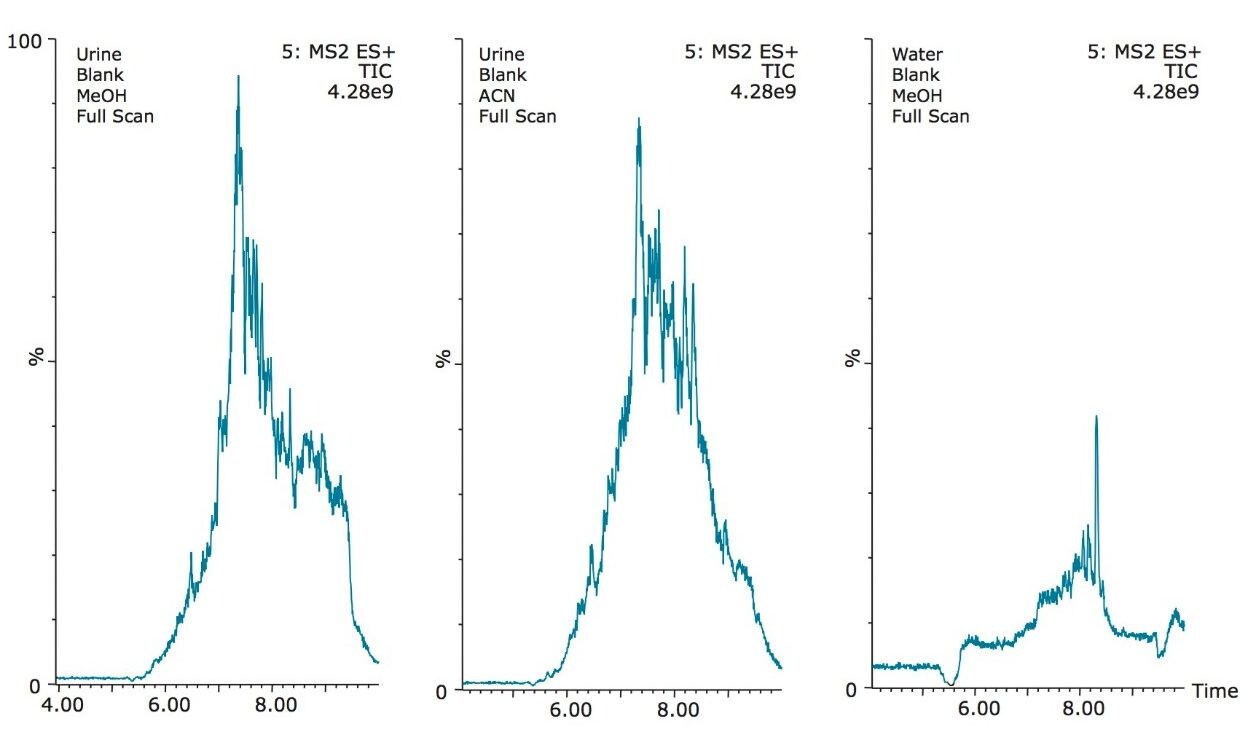
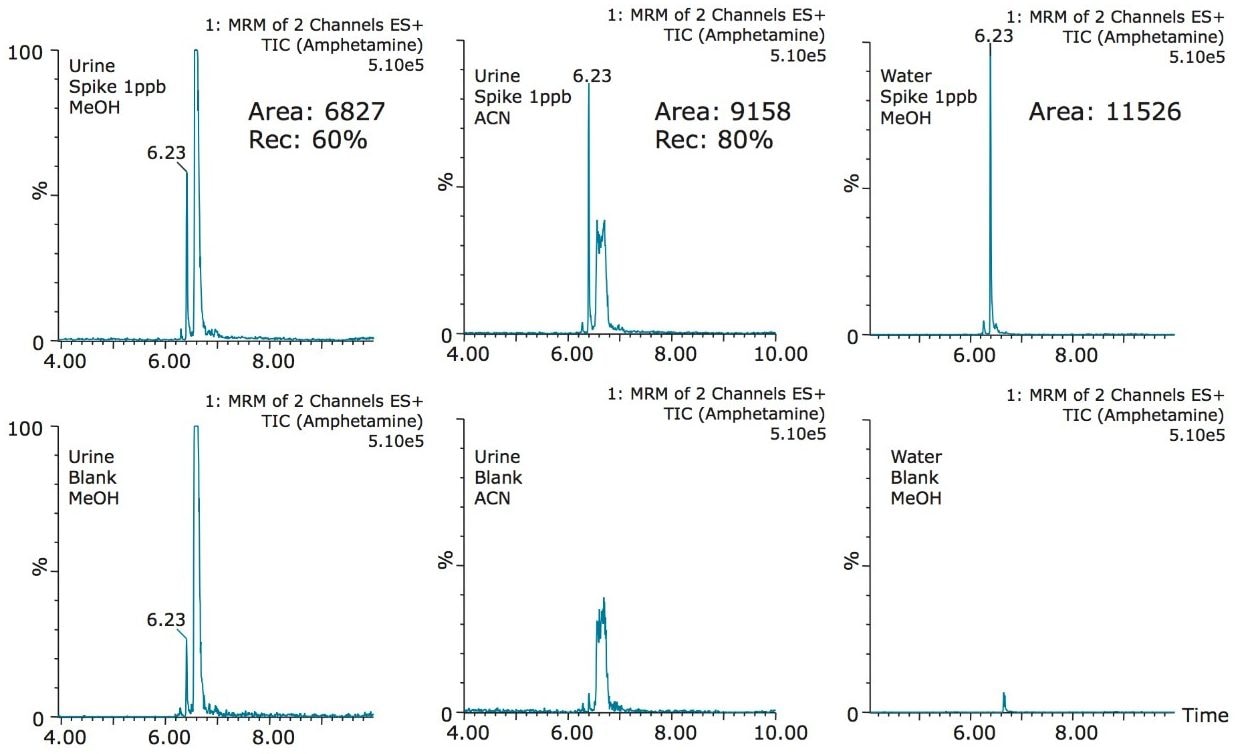
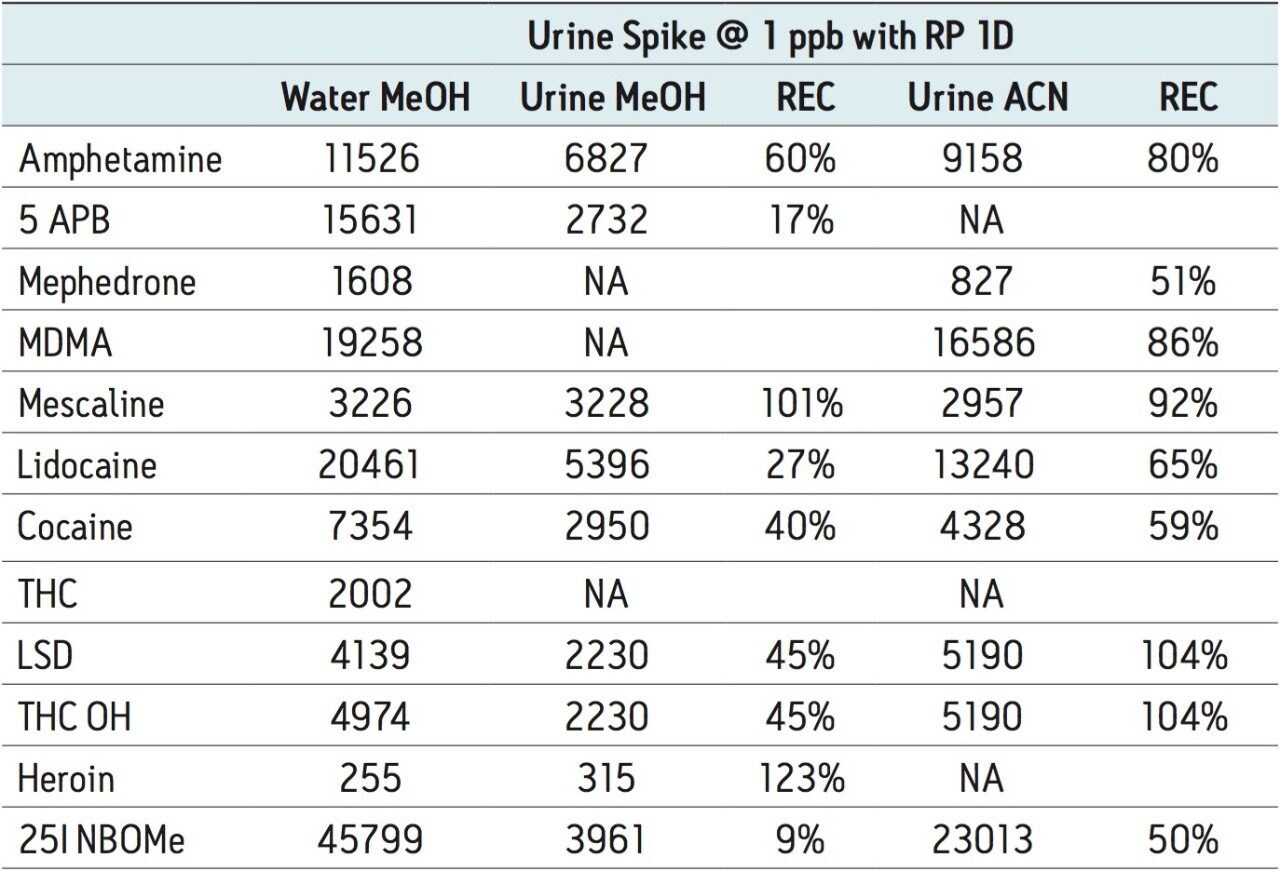
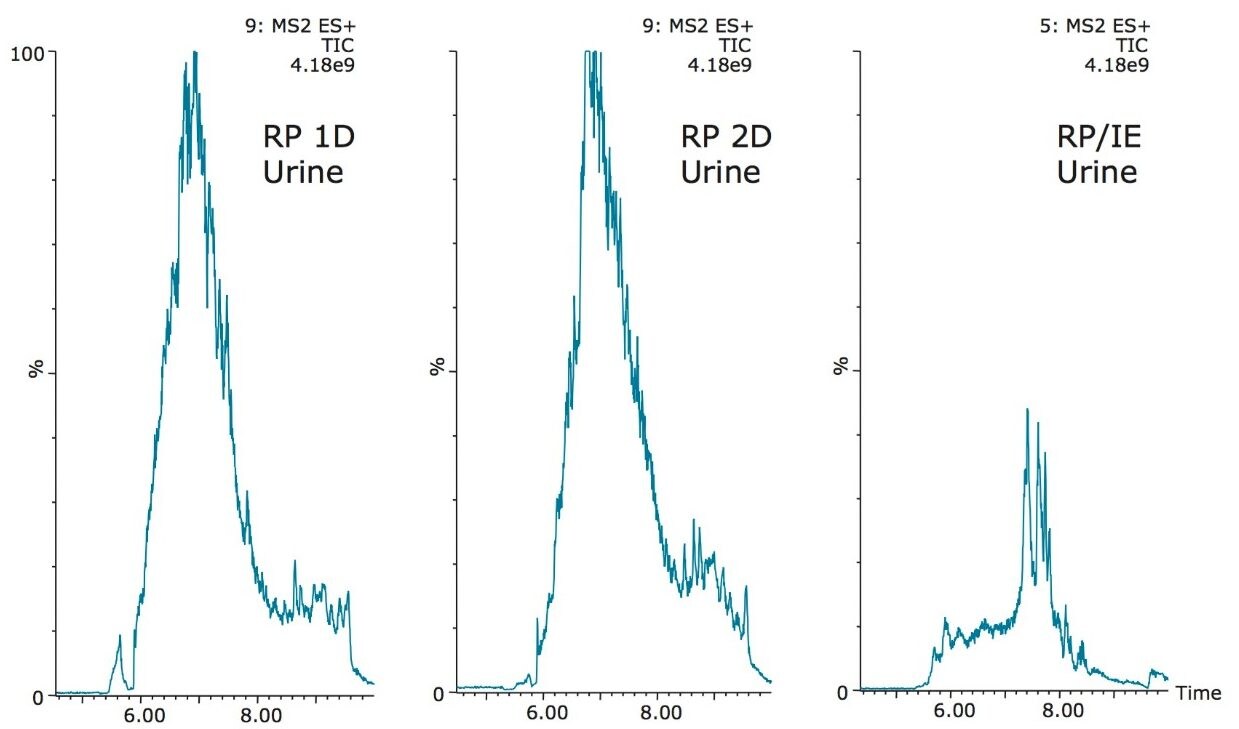
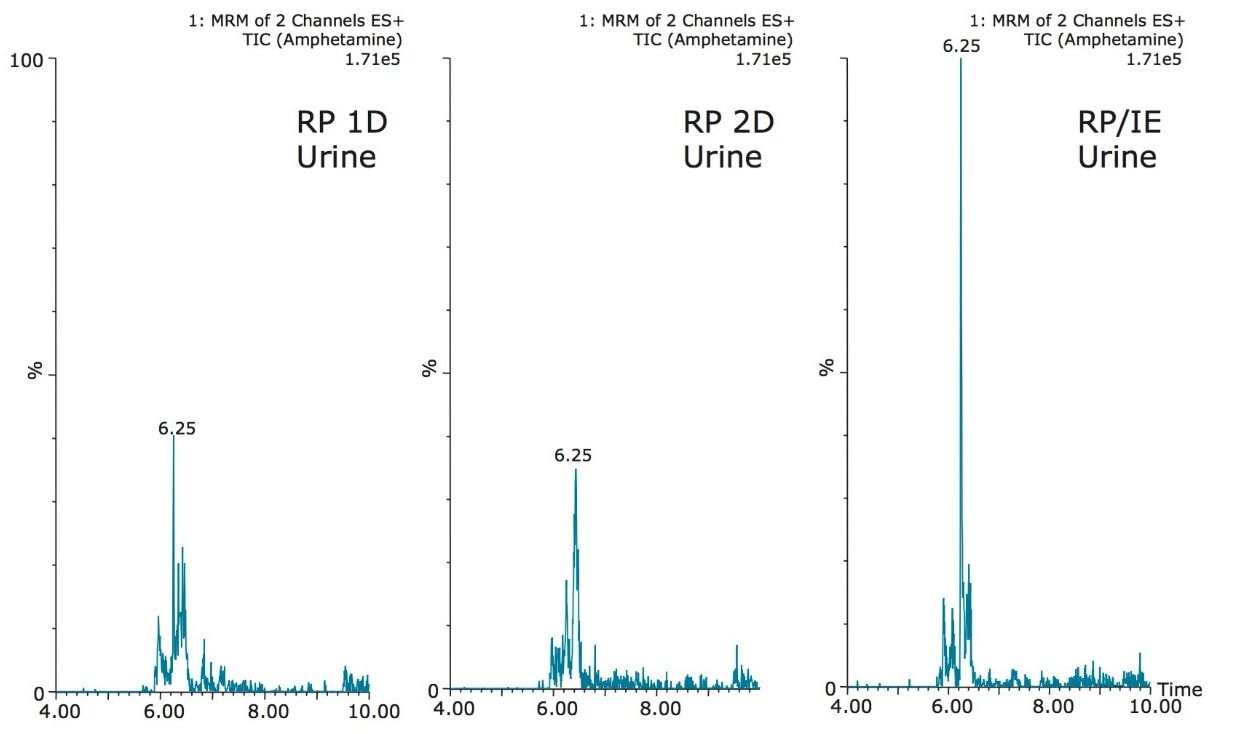
The trap and elute 2D configuration is usually constructed with two circuits. The first one is to load the analyte of interest onto a trap material and the second circuit is used for backflush elution onto a high resolution analytical column. This configuration is limited to aqueous samples only. In recent development, adding a third stream (3 pump variant) has several advantages over the two stream design.1,2 The optimization process for multi-dimensional chromatography instruments is a crucial process and can be time consuming to find optimum elution conditions. With 2D platforms, there are four key parameters to optimize that include (loading pH, trapping strength, elution strength and separation selectivity. Those parameters are split between the trap and the analytical column. For efficient trapping conditions, the loading pH, flow rate and retention chemistry are essential for ensuring a tight and narrow injection band on the trap sorbent. After the loading phase is completed, the target analytes are back flush eluted onto an analytical column for high resolution separation. For optimum separation, the usual chromatography parameters (flow rate, solvent, pH, and chemistry) are also optimized for peak performance. As such, finding the best chromatographic conditions can be quite difficult. By selecting the most common elution (Si-C4, Si-C8, Si-C18, polymer, ion-exchange... etc.) , polarity (MeOH, ACN, acetone... etc.), dilution ratio (low vs. high) and pH (low, neutral and high) well over 400 permutations can be selected. A rational approach to select the most common parameters can be autonomously screened during an overnight run (18 hours). The At-column dilution ratio is calculated from the flow rate of the loader and dilutor pump. This ratio is crucial and if not set properly can lead to peak distortion. In most instances a dilution ratio set at 5% is an excellent starting point for most analytes in methanol, acetonitrile or acetone extract (decreasing polarity value). With a dilution ratio set at 5%, the analyte (cocaine) produce a Gaussian peak shape when dissolved in methanol and acetonitrile. However, with a less polar solvent like acetone, the analyte shows signs of peak distortion (see Figure 2). The distortion can be corrected by decreasing the dilution ratio. When set at 2.5%, all three extracts show excellent peak shape. With a parallel 2D configuration, the loading phase is done on the first trap, while the second trap loaded from the previous injection is eluted toward the analytical column.