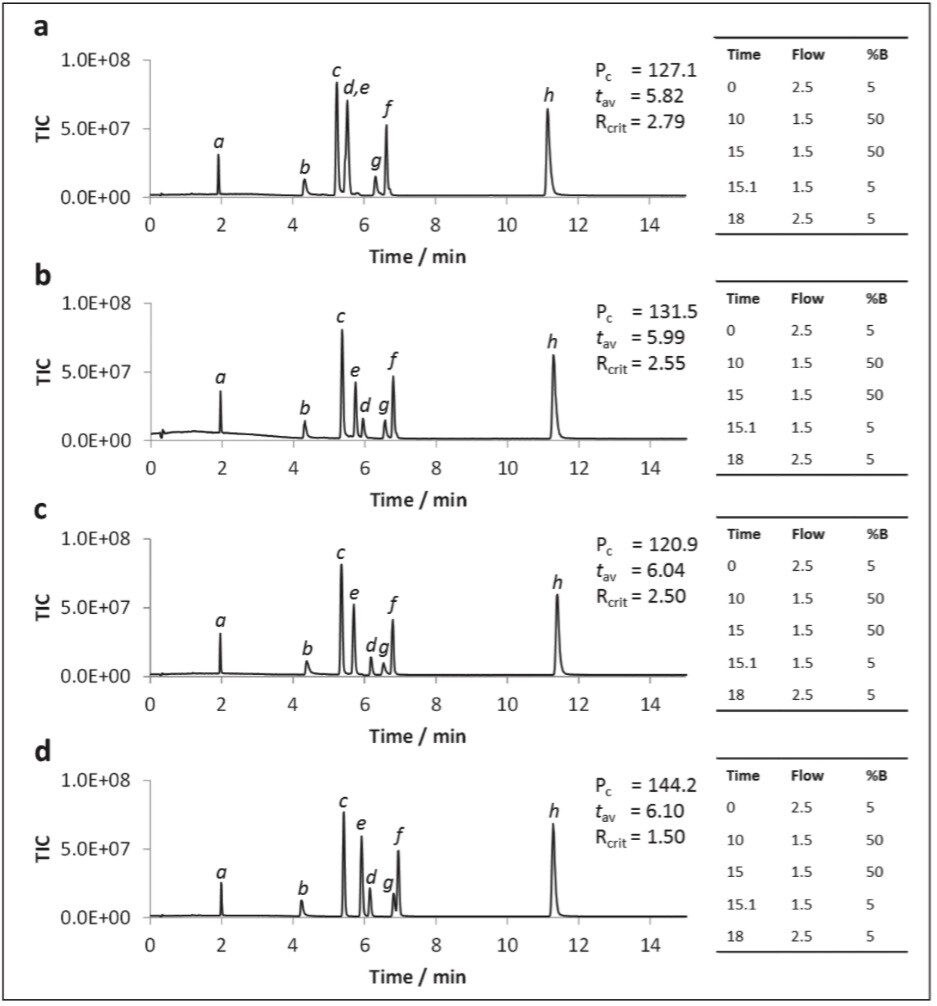
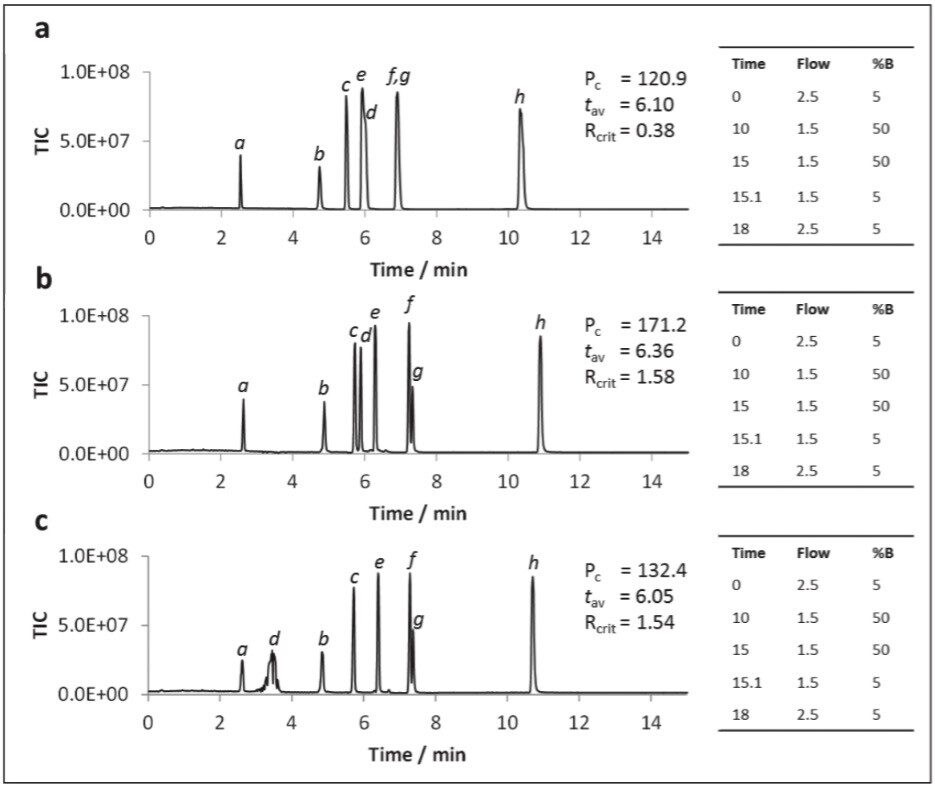
Sample description
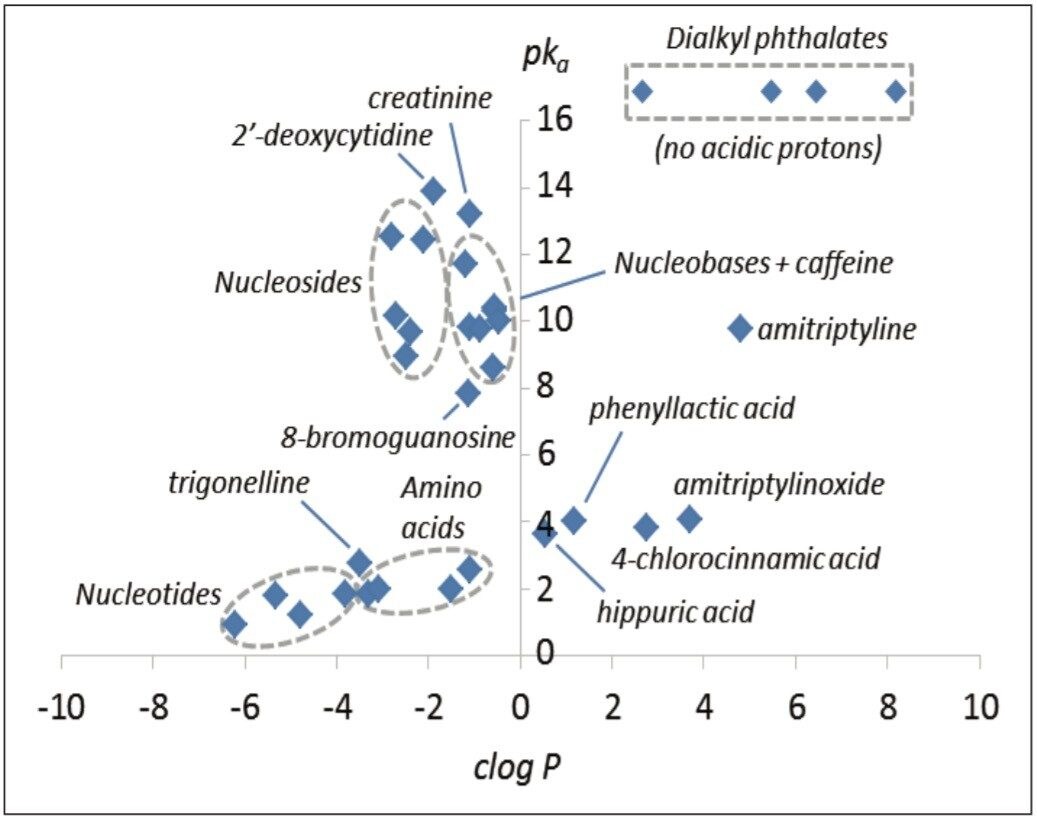
A mixture of polar analytes comprising of uracil, adenosine, creatinine, hippuric acid, cytosine, 2'-deoxycytidine, cytidine, trigonelline, caffeine, dihydrouracil, thymine, adenine, uridine, inosine, guanine, guanosine, diethyl phthalate, diamyl phthalate, dihexyl phthalate, dioctyl phthalate, amitriptyline, amitriptylinoxide, and 8-bromoguanosine was created at a concentration of 10 μg/mL-1 in water.
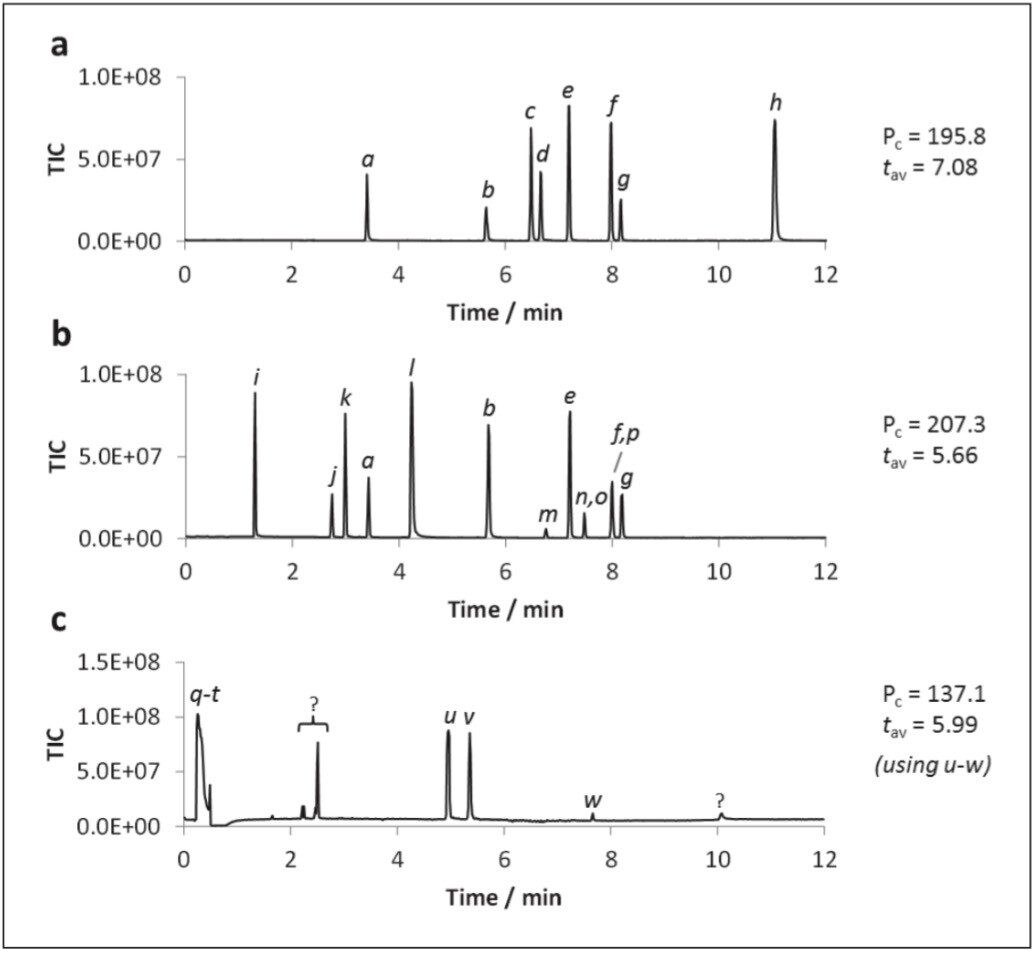
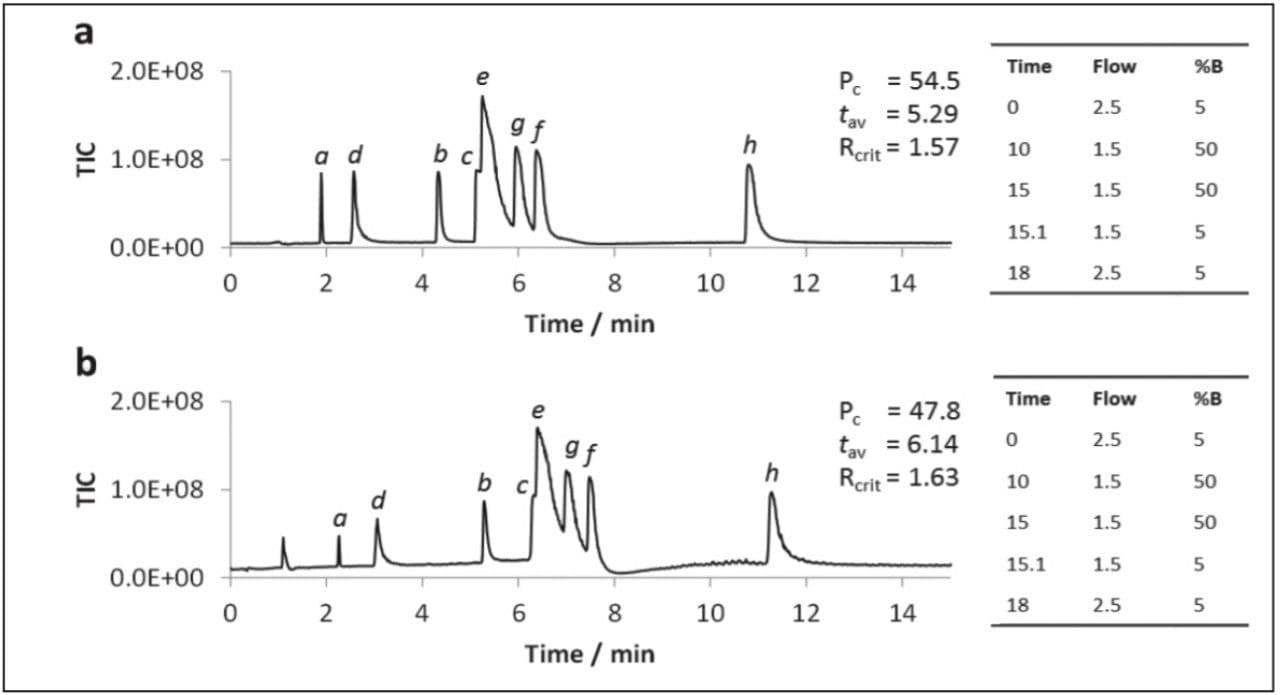
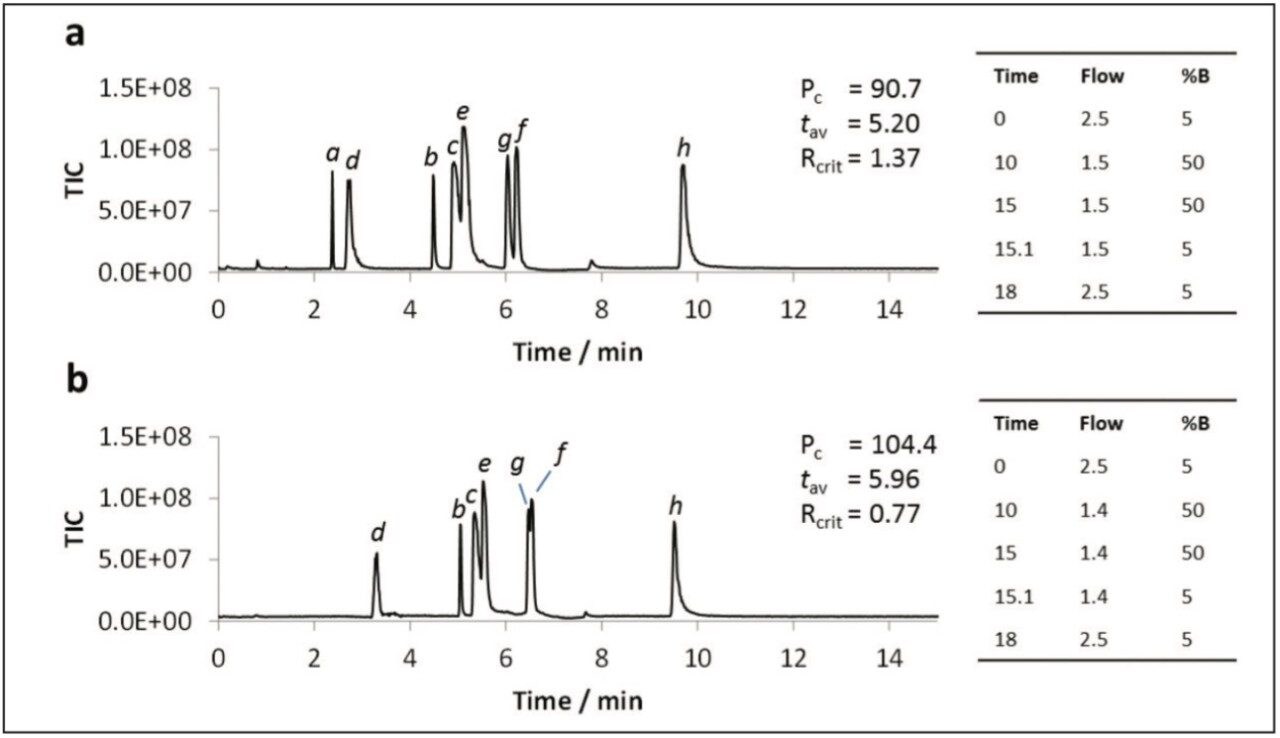
Chromatography was performed on an ACQUITY UPC2 System, comprising of an ACQUITY UPC2 Binary Solvent Manager, ACQUITY UPC2 Sample Manager, Automatic Back-Pressure Regulator (ABPR) and an ACQUITY Photodiode Array (PDA) Detector. The separation was performed on an ACQUITY UPC2 BEH Column (1.7 μm, 3.0 x 100 mm) and eluted under gradient mobile phase conditions as described in the LC conditions. Methanol was employed as the co-solvent with a starting concentration of 98:2 (v/v) CO2:MeOH, and the co-solvent concentration was then ramped up to a maximum of 60% over 12 minutes. The methanol co-solvent was modified with either water, ammonium formate buffer, or acidified buffer at various concentrations. The effluent flow rate was varied between 3 and 1.2 mL/min. The density of the subcritical fluid was regulated via a column temperature of 35 °C and ABPR pressure setting of 2000 psi. The column effluent was monitored by PDA at 273 nm or positive ion mass spectrometry.
Mass spectrometry was performed on a Waters tandem quadrupole mass spectrometer. A Waters 515 HPLC pump was employed for post column makeup flow via a splitter interface. The MS operating conditions employed for positive electrospray (ES+) of polar analytes are given in the MS conditions.