The analysis of natural and synthetic opioid drugs continues to be an important aspect of forensic toxicology. In the past, analyses were typically conducted by GC/MS after first subjecting the samples to acid or enzymatic hydrolysis to liberate glucuronide metabolites.1 With the advent of LC/MS/MS techniques, glucuronide metabolites can now be analyzed directly.2-5 Direct analyses of glucuronide metabolites can eliminate the risk of false negatives due to incomplete hydrolysis, as enzymatic efficiency can vary greatly depending upon the enzyme used and the drug substrate analyzed.6
Urine samples, unlike some other matrices, can be analyzed by “dilute and shoot” methods in which samples are diluted with an internal standard mix and directly injected onto an LC-MS/MS system.2,4 Disadvantages to this type of technique, however, include the fact that urine contains many matrix components that can interfere with MS signals. In addition, this technique does not allow for any sample concentration. This can potentially affect the quantification of some of the glucuronide metabolites that elute under high aqueous conditions, where desolvation efficiency is reduced, as well as many of the opioid drugs, since many of them do not produce intense MS/MS product fragments.
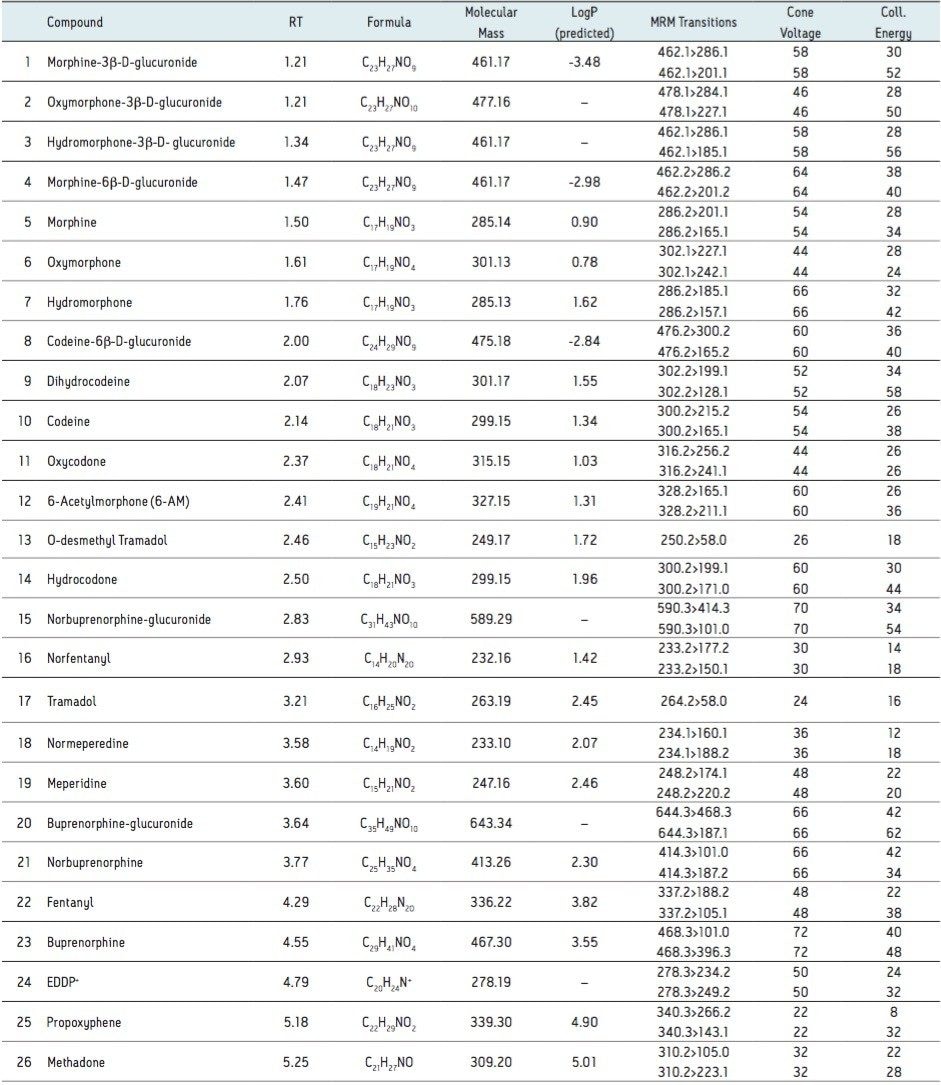
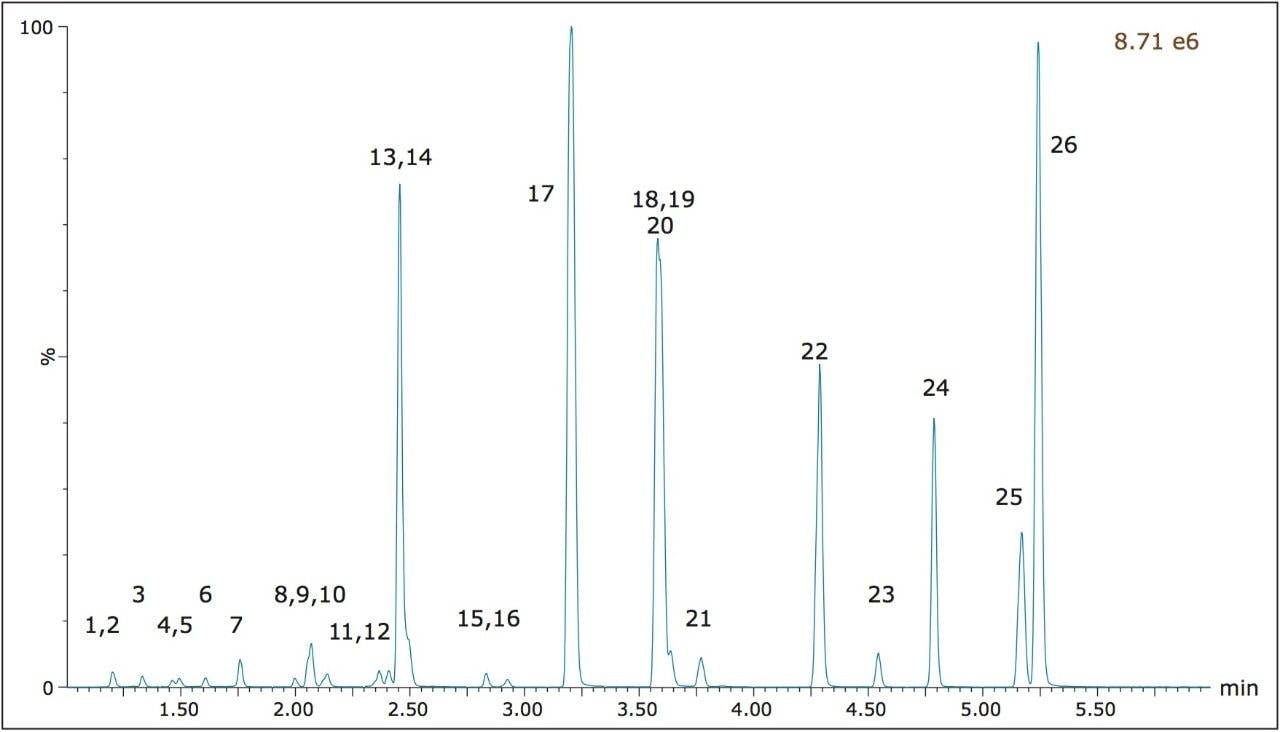
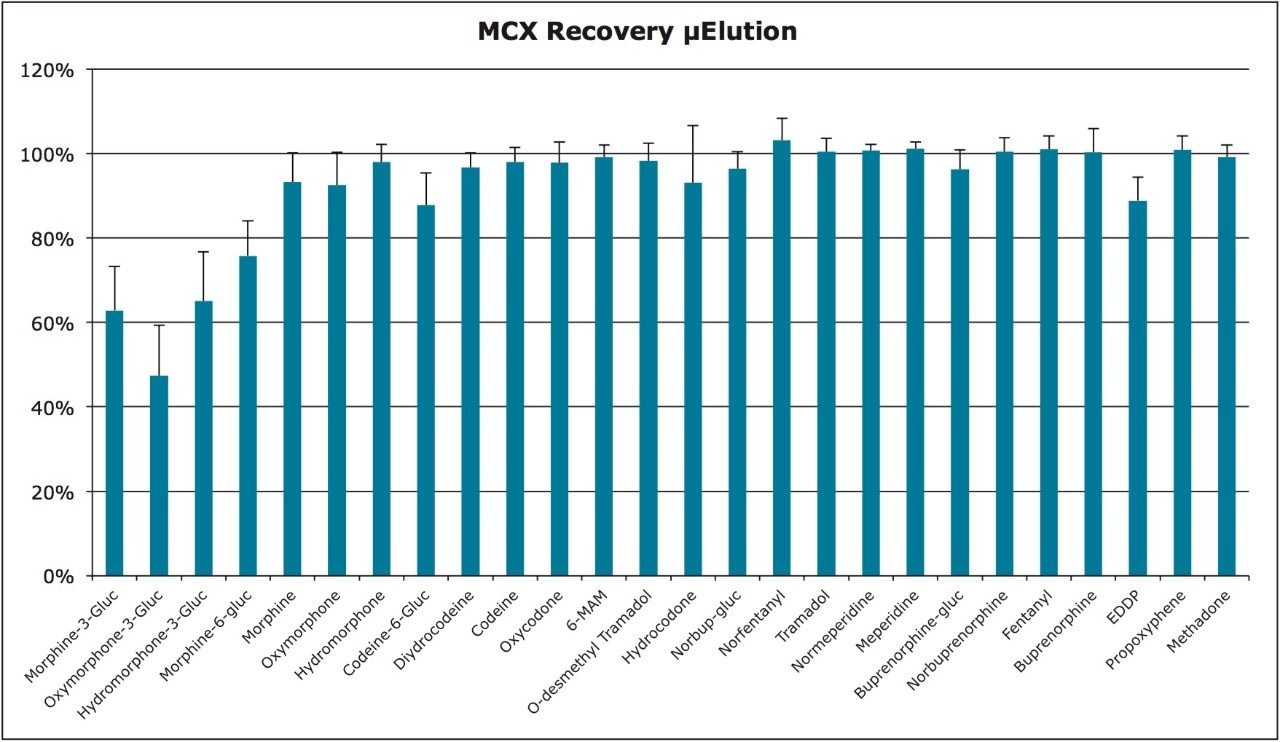
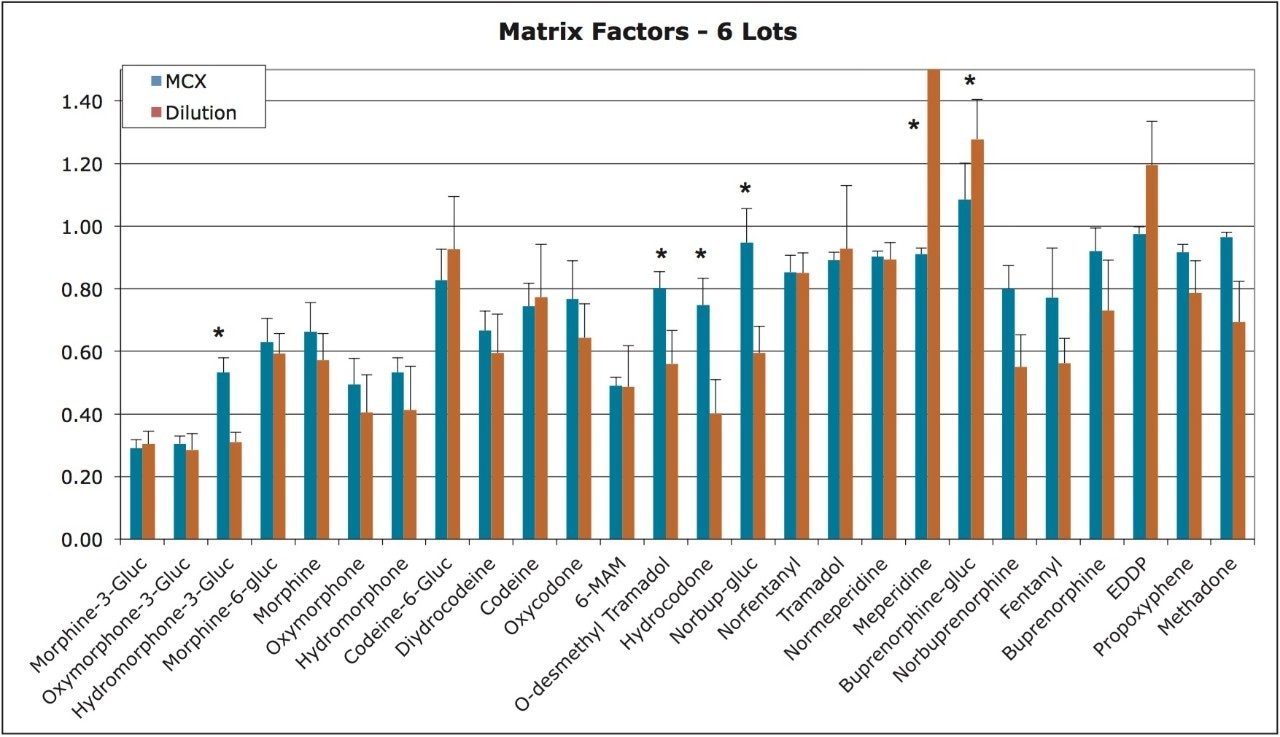
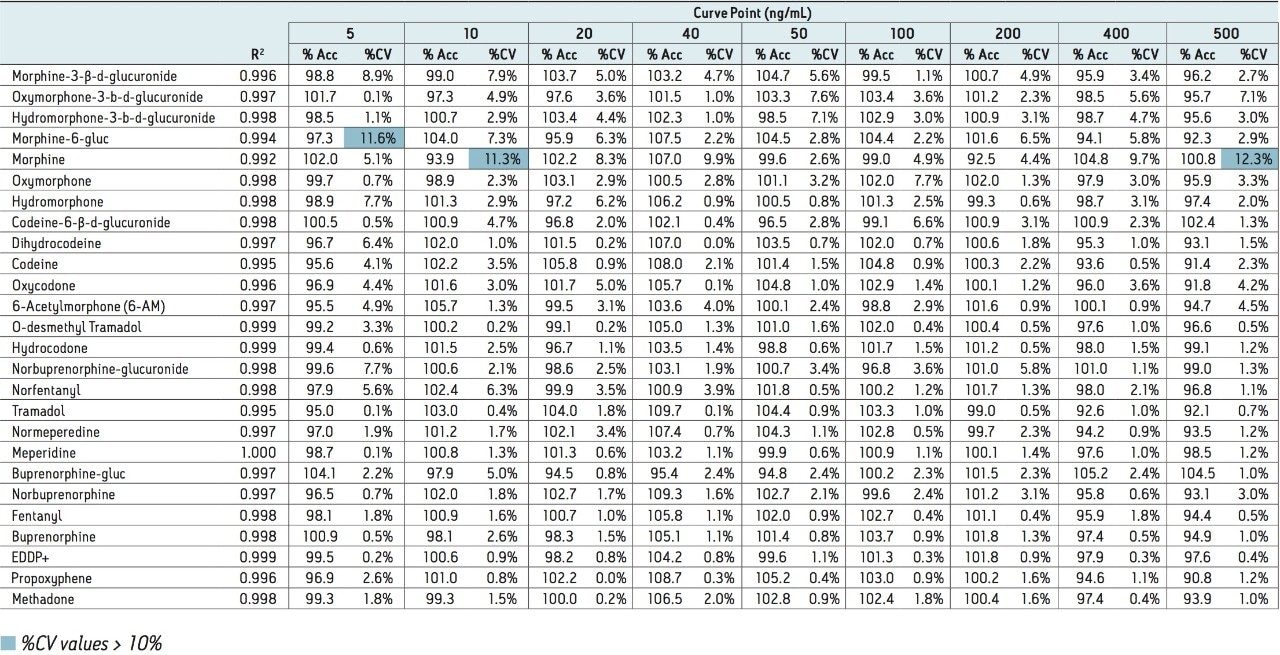
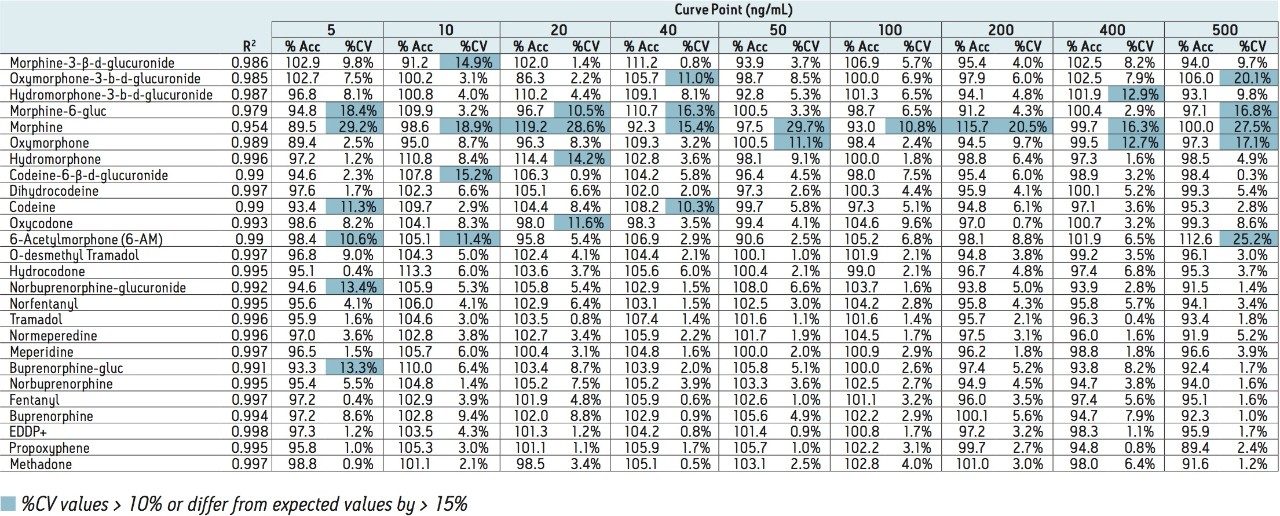
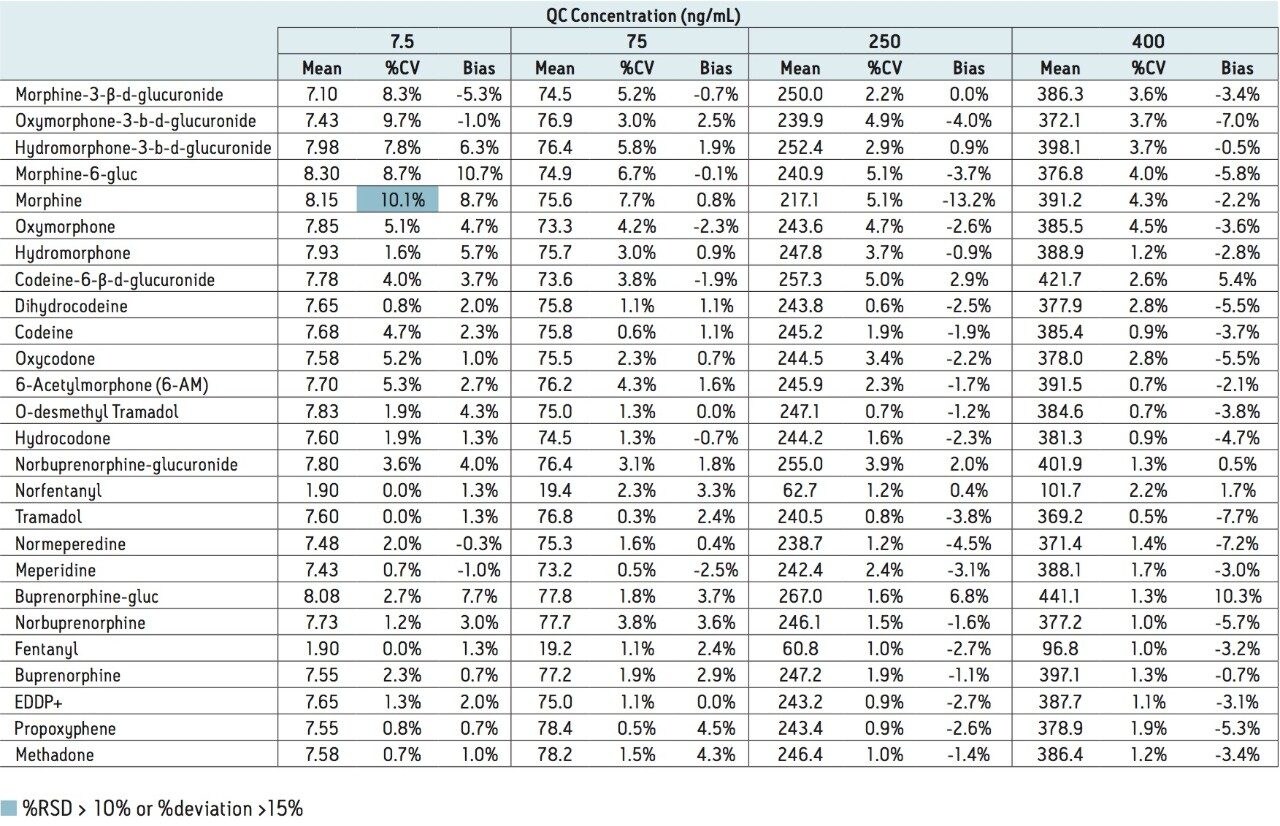
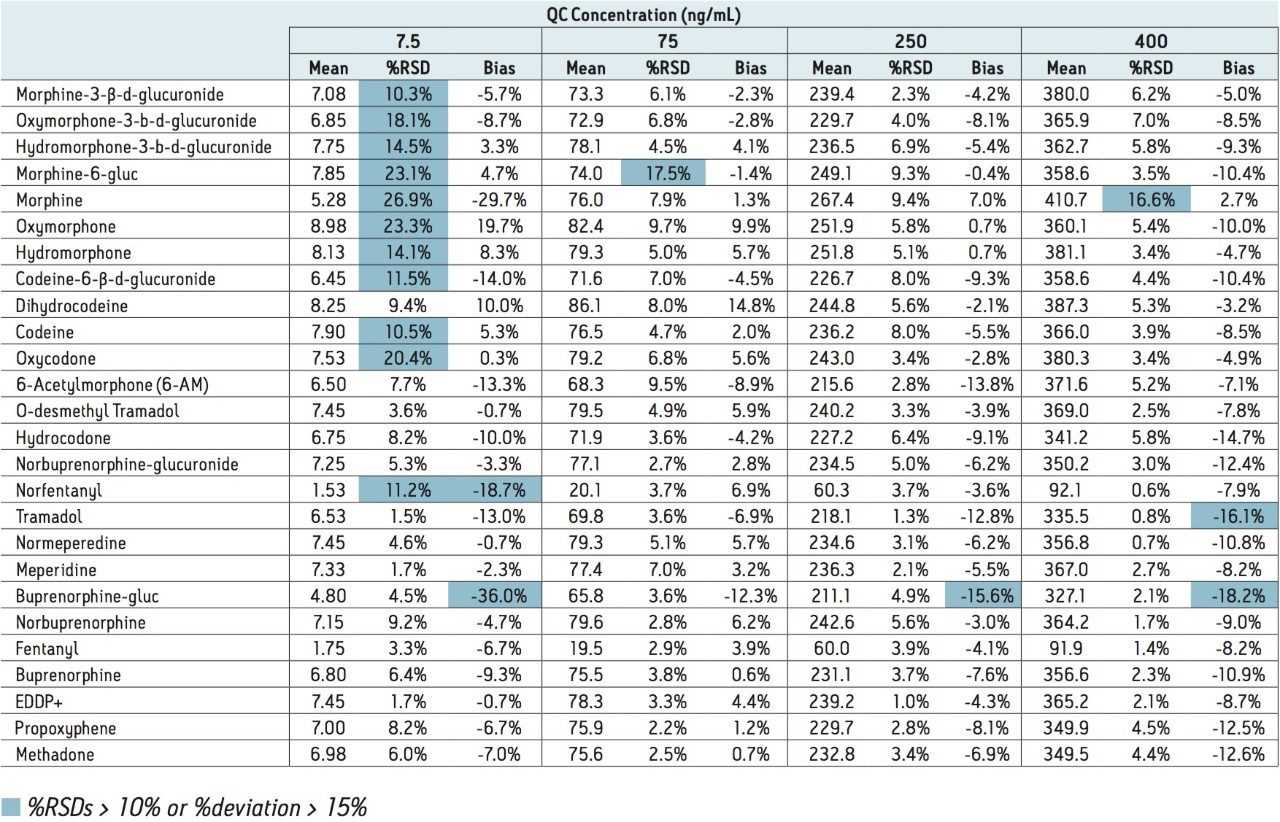
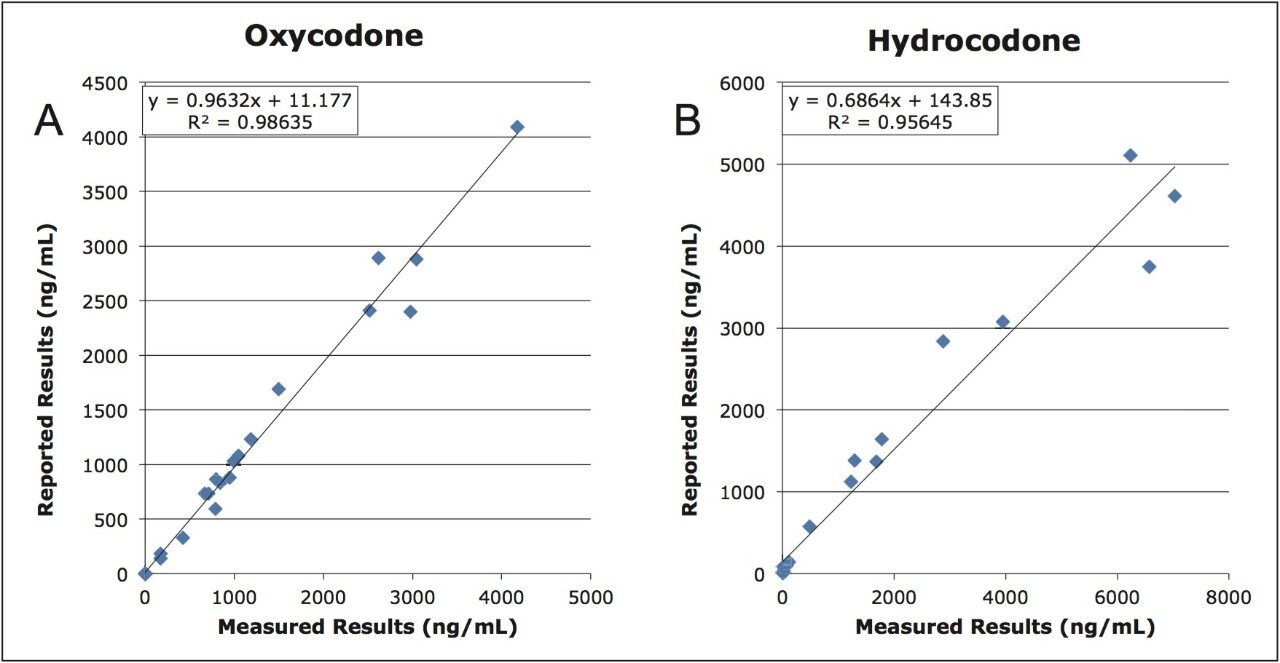
This application note highlights a method for the analysis of 26 opioid drugs and metabolites by mixed-mode SPE followed by UPLC-MS/MS. Glucuronide metabolites are directly analyzed, eliminating the need for enzymatic or chemical hydrolysis. Direct comparison demonstrates that mixed-mode SPE has improved linearity, greater accuracy and precision, and fewer matrix effects than a simple dilute and shoot method. Previously confirmed, incurred samples were also analyzed, allowing for additional evaluation of this method.