Within- and between-batch imprecision determined by replicate extraction and analysis over 5 days of a human plasma base pool spiked to 3 concentrations revealed mean within batch imprecision of <7.6 and <5.6 % coefficient of variation (CV) for M and NM, respectively. The same data reveals the mean between batch CV as <7.5 and <12.8 % for M and NM, respectively.
Linearity of detector response up to 24.55 nmol/L was demonstrated with the 8-point calibration curve showing a coefficient of determination >0.995 and <15 % deviation from nominal concentrations. Additionally, linearity of detector response upon extraction of an 8 nmol/L spiked sample, sequentially diluted with stripped serum, was shown.
Mean Oasis WCX µElution plate SPE efficiency was calculated as the peak area ratio of pre- to post-extraction spiked plasma (n=3) was 97 and 95 % for M and NM, respectively. Matrix effects were evaluated from the peak area of the post-extraction spiked samples (n=6) taken as a percentage of extraction solvent spiked to equivalent concentrations. Mean (range) matrix effects were 16.4 (1.8 % to 35.0) % ion enhancement for M and 8.7 (3.6 % suppression to 19.4) % enhancement for NM. Calculations using analyte: internal standard peak area response indicated matrix effect compensation by the internal standard with a mean net matrix effect (range) of 4.0 (0.5 to 6.0) % enhancement for M and 0.3 (3.3 % suppression to 7.5) % enhancement for NM.
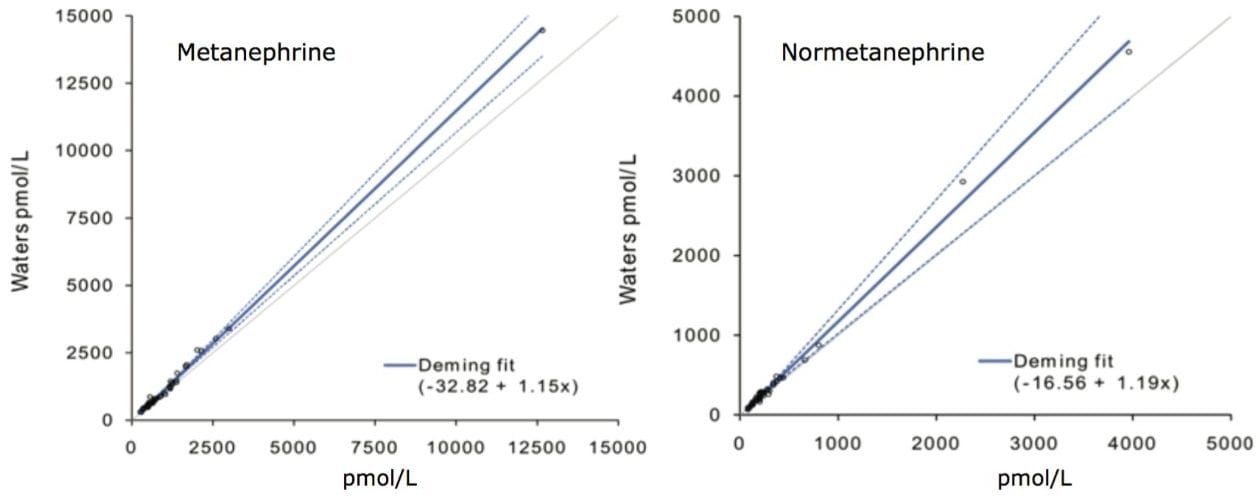
Comparison was made using 50 plasma reference samples analysed by an online SPE LC-MS/MS method.2 Deming regression conducted with Analyse-it for Microsoft Excel for Windows 2003 showed neither significant proportional nor constant bias across the measured range of 87–4455 pmol/L for M (p>0.05). Proportional bias of 15 % was found across the measured range of NM 300–14459 pmol/L (p<0.02), however, this became insignificant when limiting the comparison to below the upper limit of the reference sample interval (<1070 pmol/L; p=0.48).