In the field of bioanalysis, the analysis of a target analyte and its metabolites in biological fluids (urine, plasma, and blood) is predominantly the main activity during animal and human trials. As such, a wide range of extraction and analytical techniques are used for qualitative and quantitative analysis. For accurate quantification, the target analyte must be isolated from the matrix,1,2 from which a clean and concentrated extract can be analyzed by GC-MS or LC-MS for trace level detection (ng/mL).
During method development, it is quite common to include an enrichment step if the analytical protocol requires sub-level (ppb) detection. Therefore, large sample quantities are required during the extraction process. For human and large animal trials, adequate sample size (volume or mass) can be made available for trace level analysis. However, for pre-clinical trials, sample volume drawn from small rodents (rats, mice, guinea pigs, etc.) is significantly lower and can limit the sampling frequency. As a result, the concept of “micro-sampling” has gained an interest as a potential alternative. From a logistical standpoint, handling and shipping of liquid samples adds an additional level of difficulty. As such, the concept of “Dried Blood Spot” (DBS) allows the collection of micro– (<200 μL) sampling directly onto a sorbent support card. After an adequate air drying period, the dried sample can be stored and shipped under ambient temperatures. Once the DBS card has reach the laboratory, the challenge now resides in the isolation of the target analyte from the dried matrix.
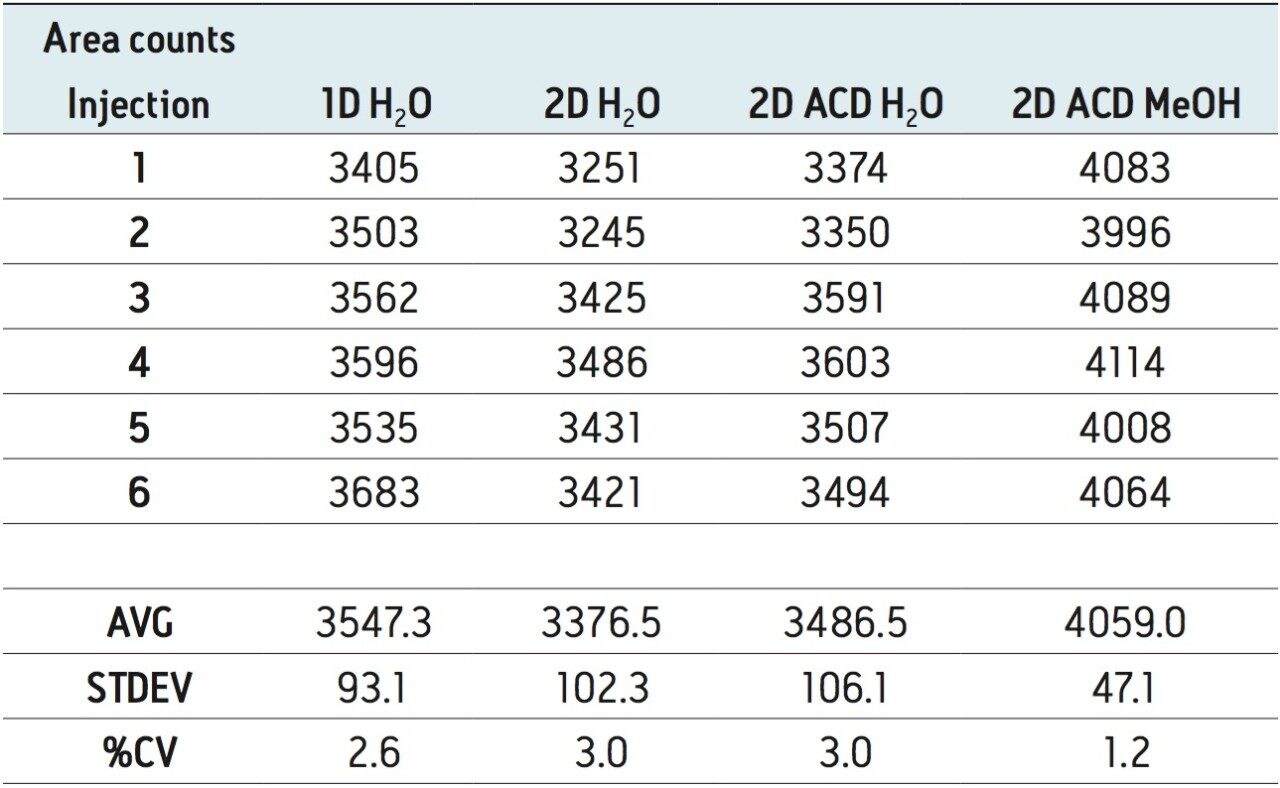
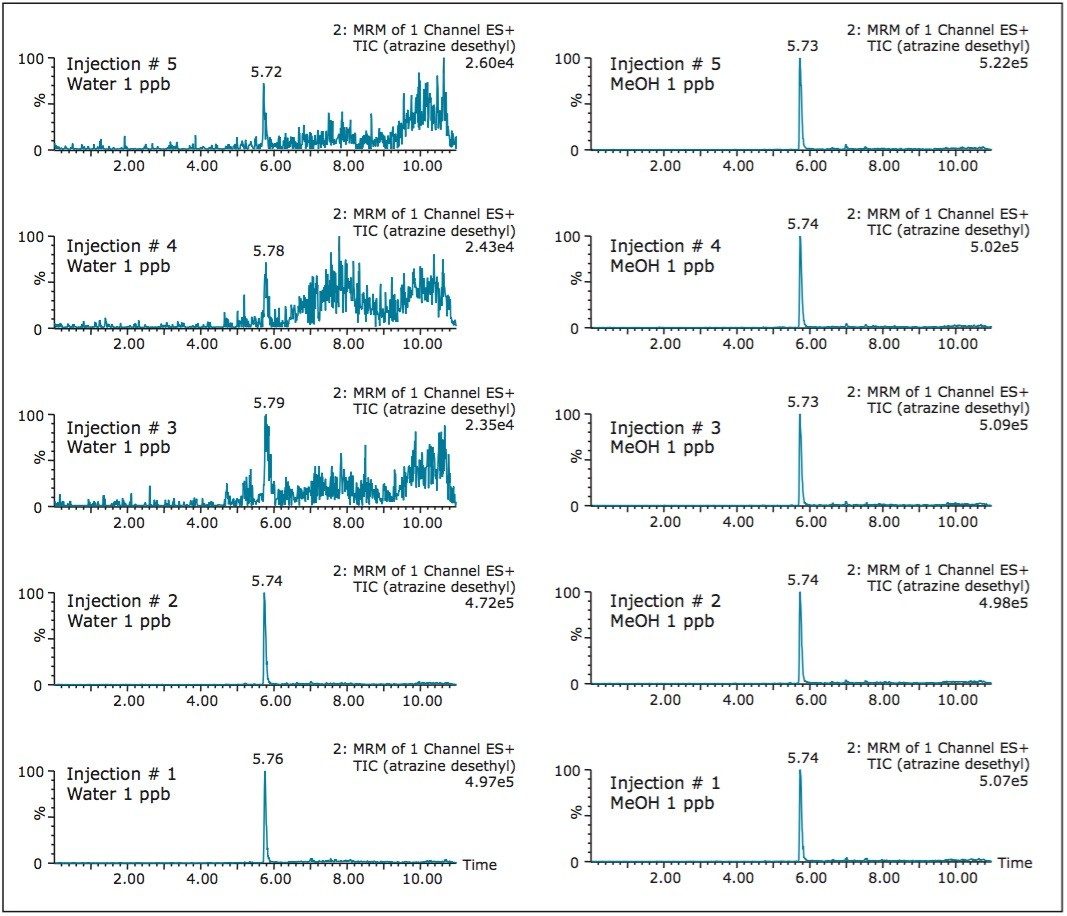
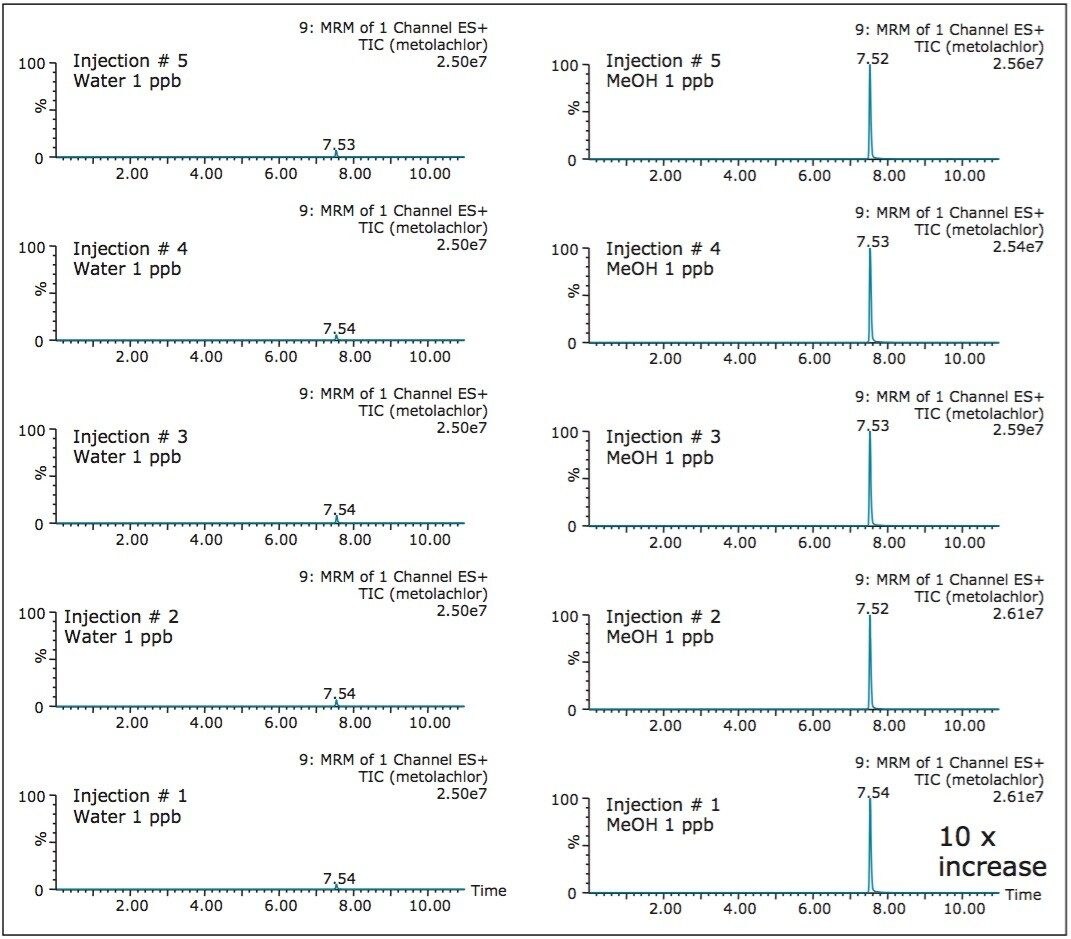
The extraction procedure of a DBS card requires the removal of bounded materials from the support structure, most often cellulose, using a punch cutting technique. The punch cutting technique utilizes a circular cutter to cut a fixed surface of the DBS card. The round disk is then subjected to a solid-liquid extraction with water or organic solvents (IPA, MeOH, ACN, etc.). With aqueous extraction, if the extract is of good quality, a small aliquot (10 μL) is used for analysis by LC-MS/MS. However, in most instances, the extraction efficiency gives better recoveries with organic solvents.
As a consequence, a solvent conversion step must be added to the extraction protocol. This is achieved by removing the organic solvent using an evaporation and reconstitution step. The evaporation by nitrogen stream is the most popular method, but not without difficulties: it is a well know fact that evaporative loss can occur and create a potential cause for poor recoveries.
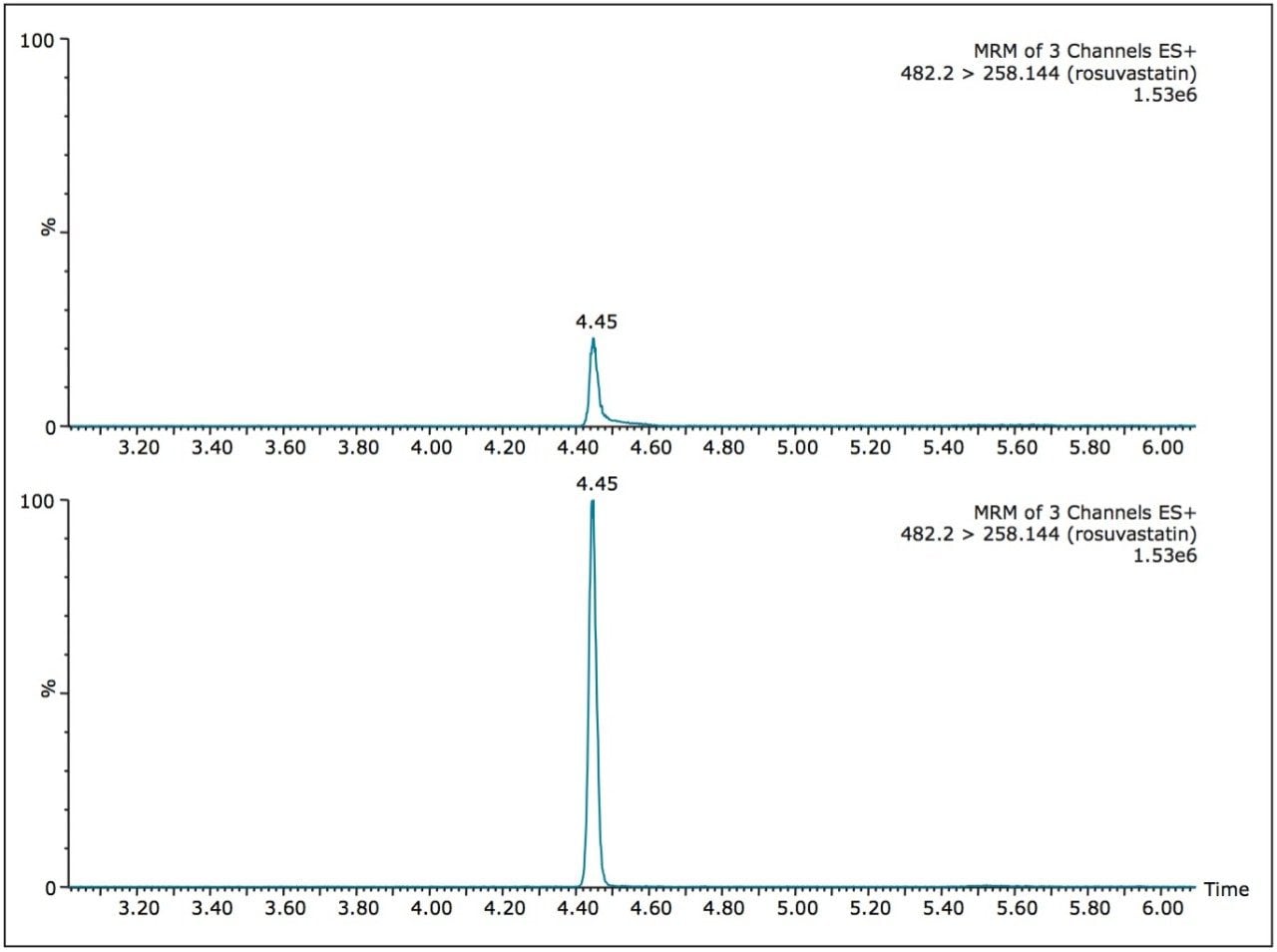
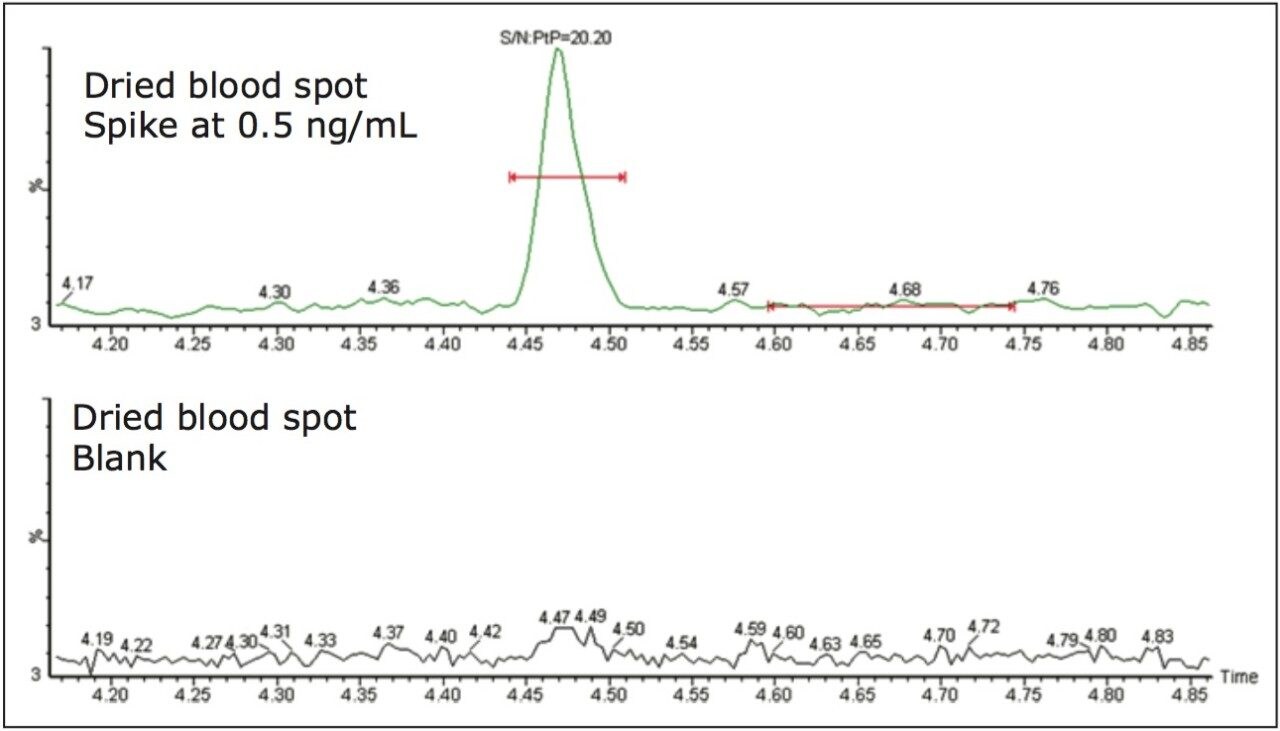
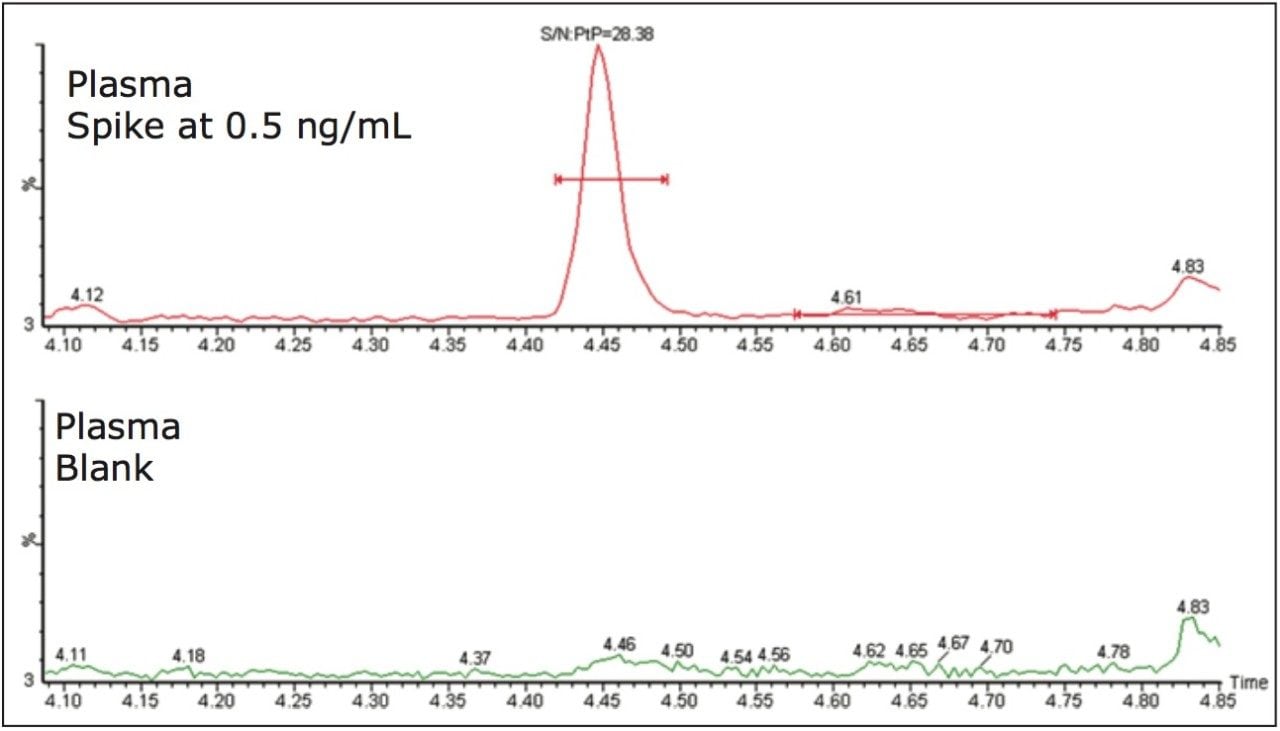
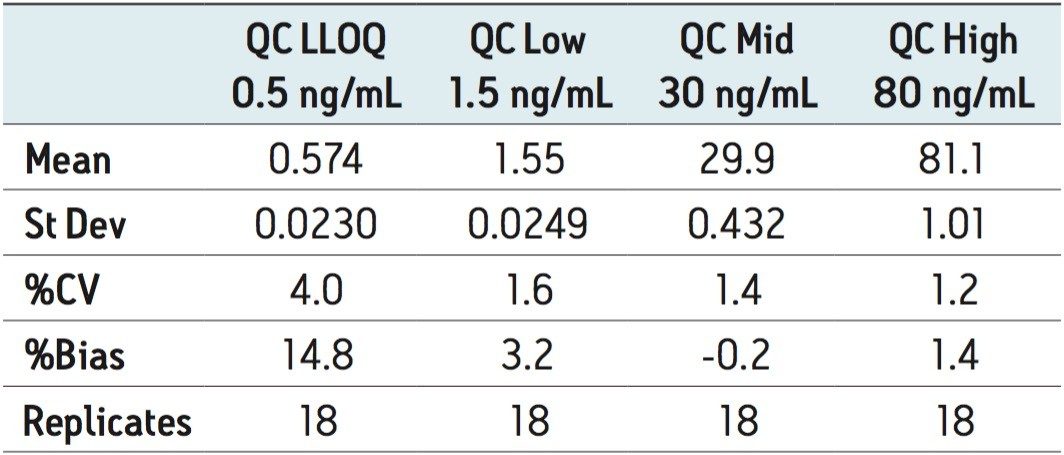
ACQUITY UPLC with 2D-LC technology3 allows the option of large-volume injection (up to 1000 μL) of aqueous and organic extracts, thus removing all evaporation and reconstitution steps from any extraction protocols. In this application, 15-μL aliquots of blood were spotted onto Whatman DMPK type B support cards. A 3-mm punch disk was suspended in 100 μL methanol. The LC-MS/MS analysis of rosuvastation was performed by using an injection volume of 85 μL enabling a detection limit of 0.5 ng/mL.