Three replicates of two Escherichia coli samples, differentially spiked with bovine serum albumin, alcohol dehydrogenase, enolase, and glycogen phosphorylase B were analyzed. The injected on-column amounts for the spiked proteins in the first sample were 500 attomoles each and 4,000, 500, 1,000, and 250 attomoles for the second sample, respectively. The peptides were separated and analyzed using a Waters nanoACQUITY UPLC System coupled with a SYNAPT G2-S operating at a precursor and product ion mass resolution of > 20,000 FWHM, with data acquired in LC-MSE scanning mode. Searches were conducted with ProteinLynx GlobalSERVER v.2.5.2 and quantified with Progenesis LC-MS.
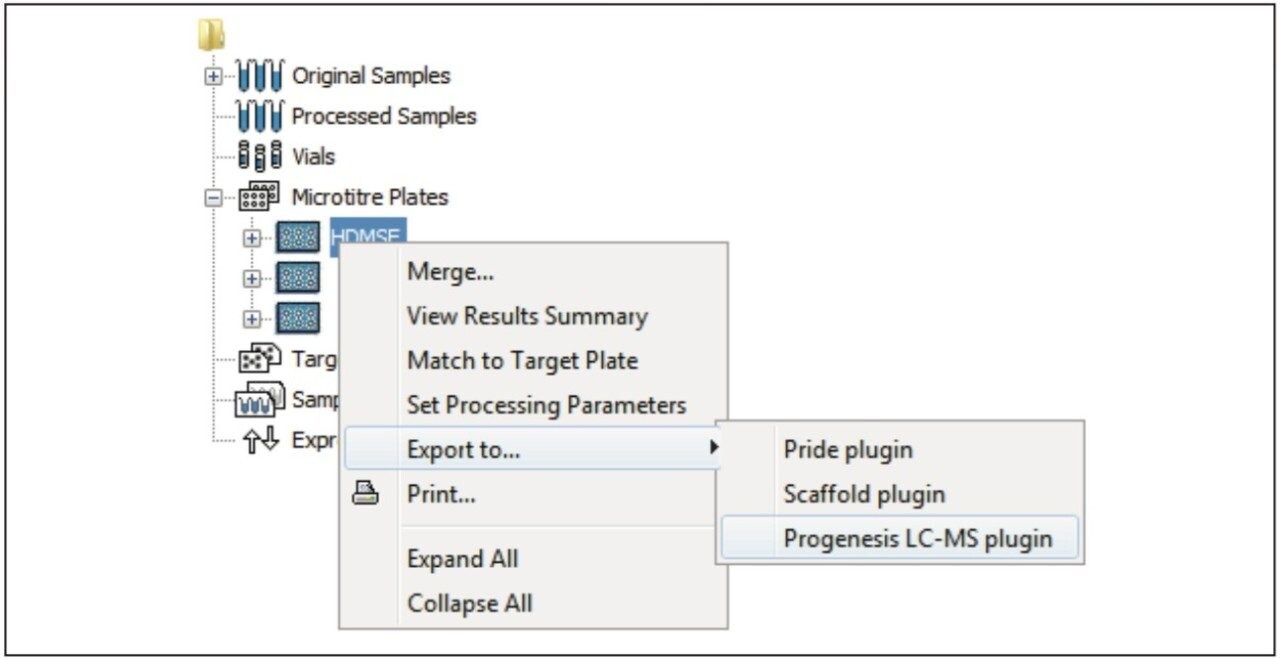
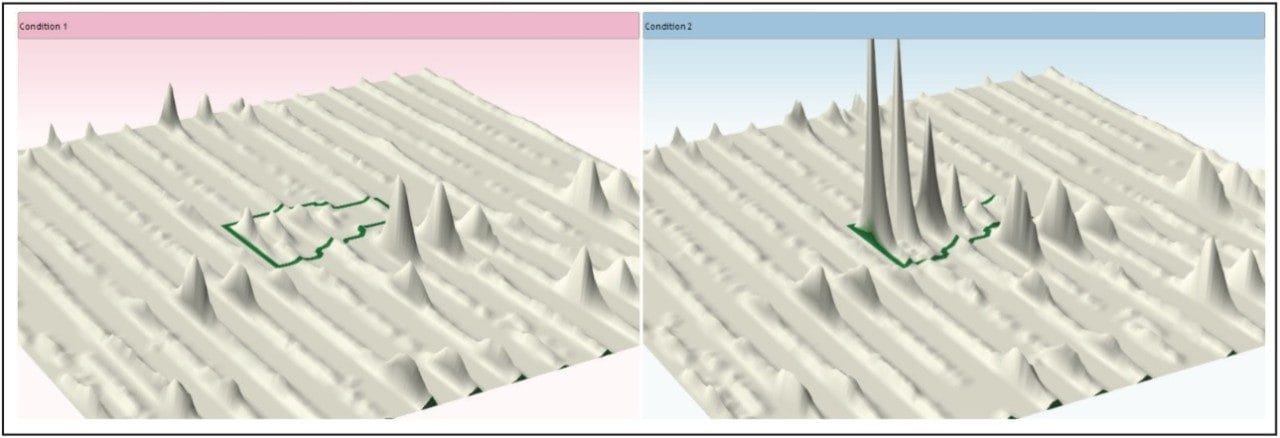
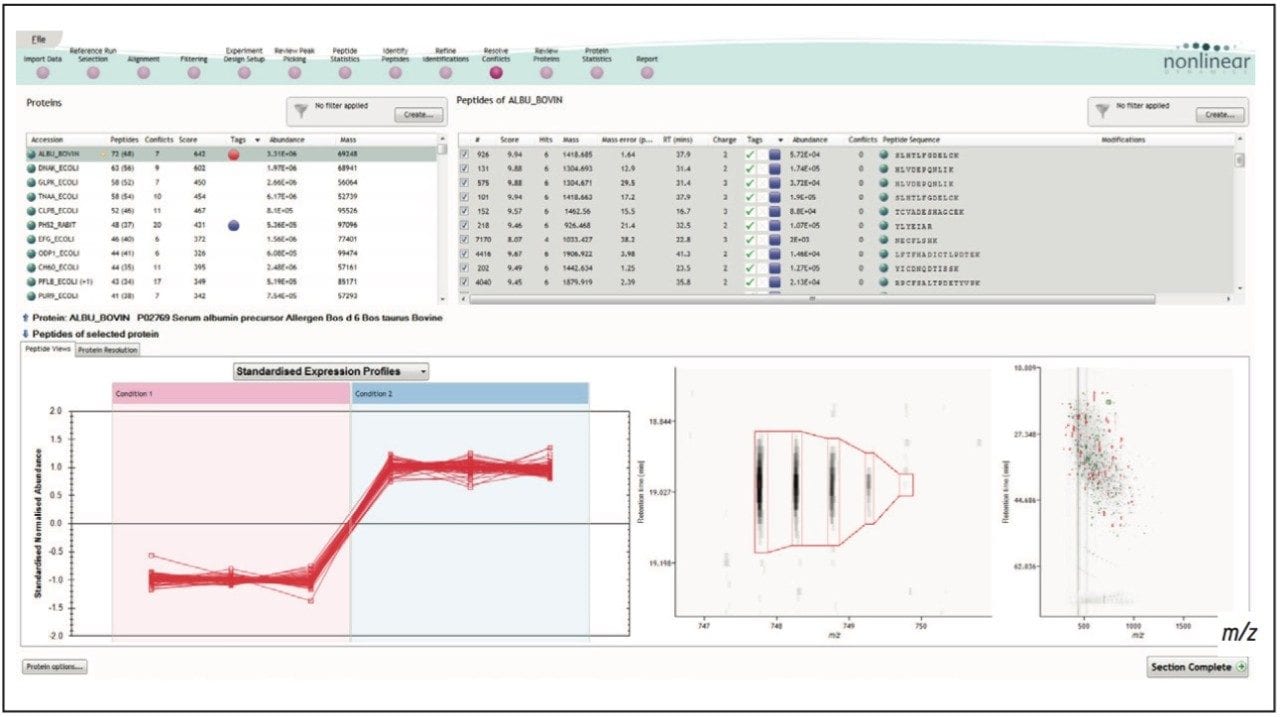
Figure 1 illustrates the plug-in export tool of ProteinLynx GlobalSERVER, which is capable of automatically creating one or more Progenesis LC-MS compatible data files. Visualization of the raw data in 3D-montage view is shown in Figure 2, which confirmed the increased on-column of one of the proteins in the second sample. In this instance, one of the isotopic distributions for one of the charge states of an up-regulated bovine serum albumin peptide is shown. A protein centric summary is shown in Figure 3, which details the peptides of the selected protein, a normalized regulation distribution for the six samples analyzed, a 2D-montage view of the aggregate run, and a fullrange contour LC-MS image of the latter highlighting the peptides of interest.