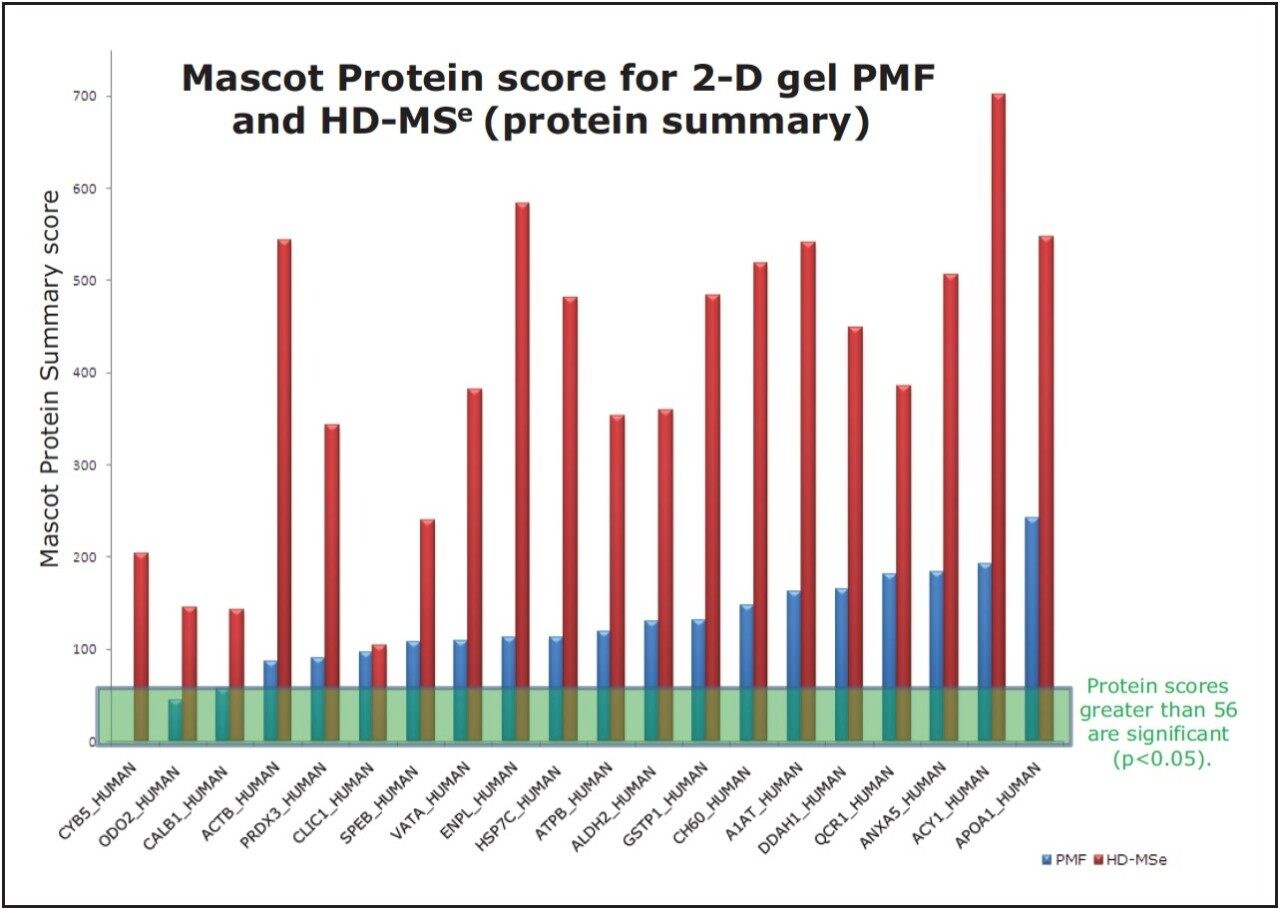
Peptide Mass Fingerprinting (PMF) is an unbiased protein identification technique that offers limited sequence information for identification purposes due to the reliance on the m/z measurement of proteolytic peptides.
An alternative technique commonly used conducts MS/MS sequencing experiments by fragmenting the tryptic peptides through sequential selection using the first analyzer of a mass spectrometer. This MS/MS approach has the following limitations:
- Only the most intense peptides are fragmented
- The user must choose the peptide to fragment
- Information is limited, as peptides can be missed if the sample is depleted
- After the sample is discarded, regaining knowledge from the sample is not feasible
- Potentially more time consuming than PMF alone
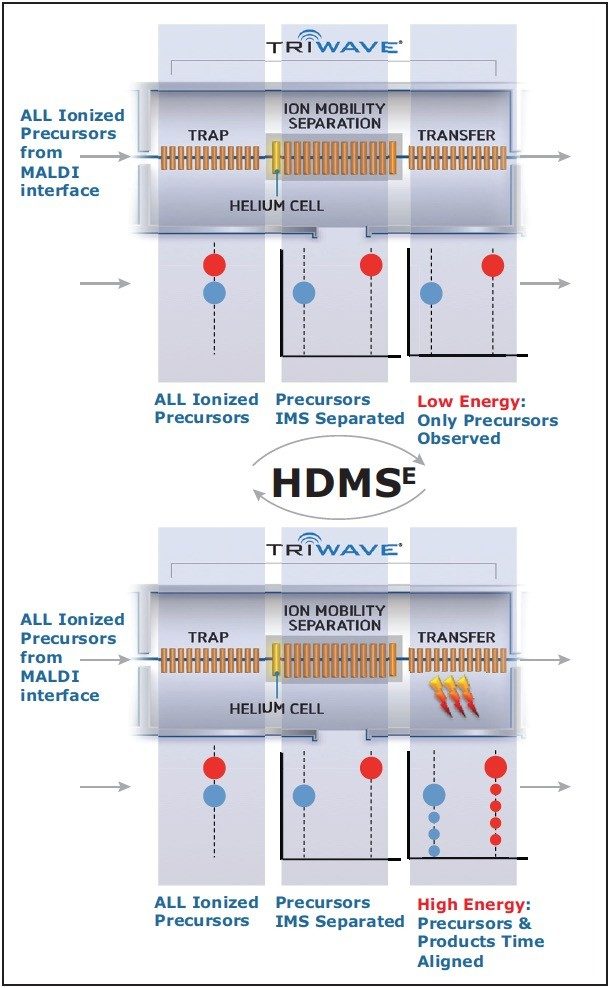
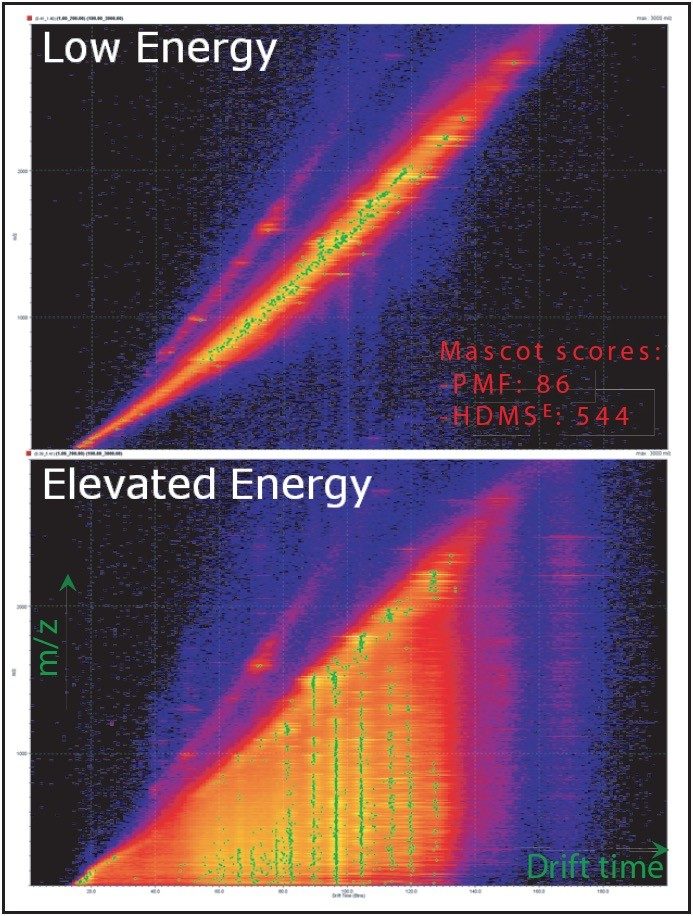
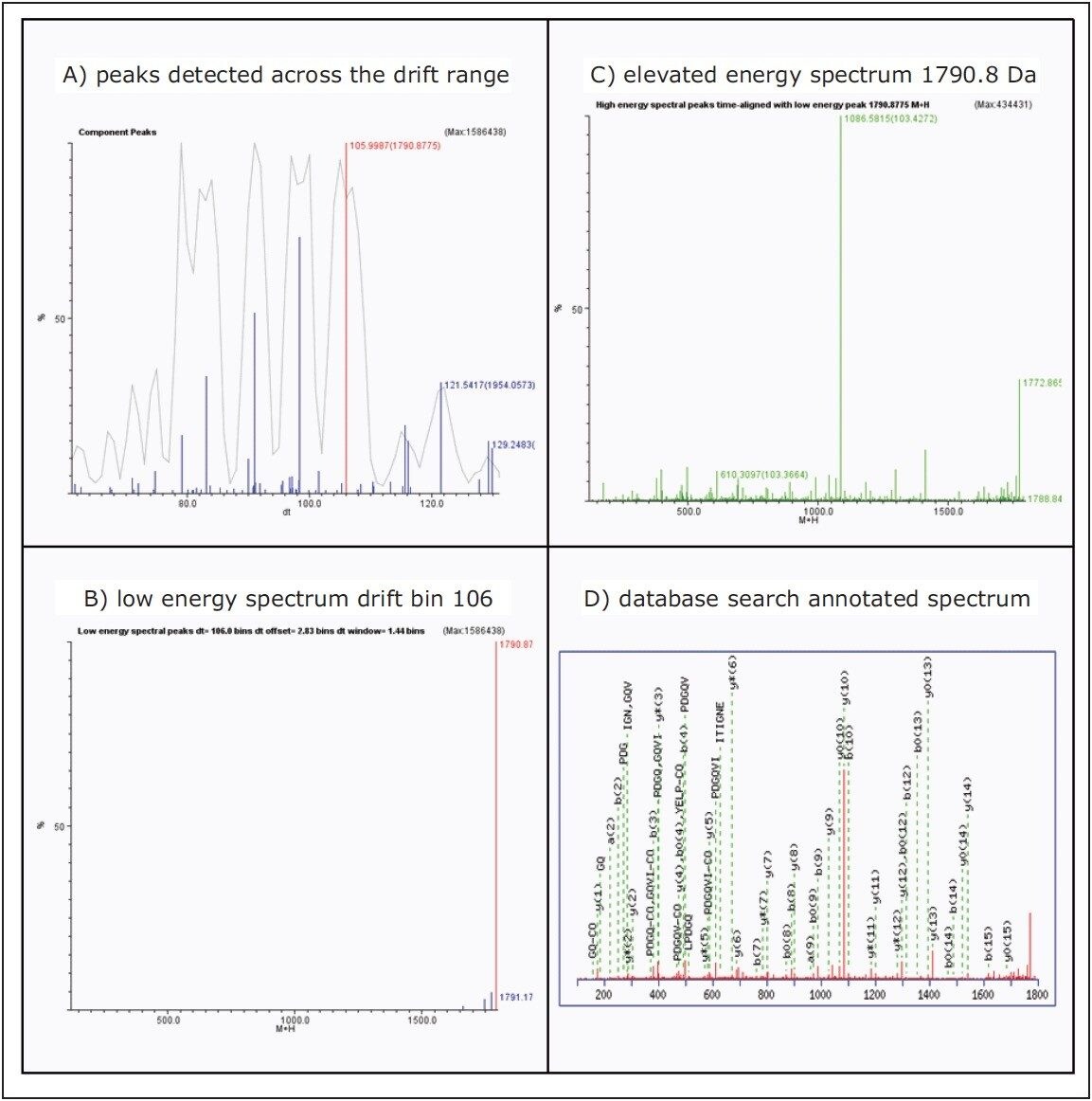
In this application note, we propose a novel method of acquiring data called High Definition MSE (HDMSE). This method maintains the independent nature of PMF but extends its utility by providing molecular and MS/MS-like peptide sequence information in a simple, generic fashion. Unlike traditional MS/MS analyses, where selection of the precursor ions by the first mass analyser is used, ion mobility separation (IMS) is utilized as an orthogonal separation of peptides in the gas phase, followed by simultaneous fragmentation and high resolution mass analysis. Peptides are associated with their fragments for protein identification using ion mobility (drift time) alignment.