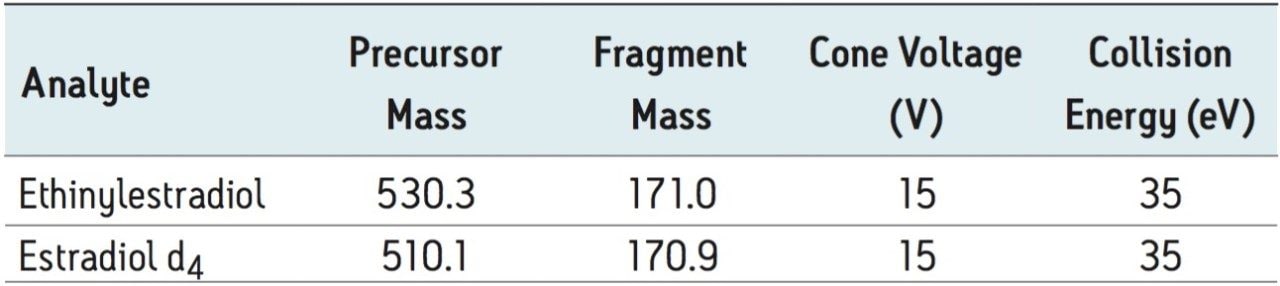
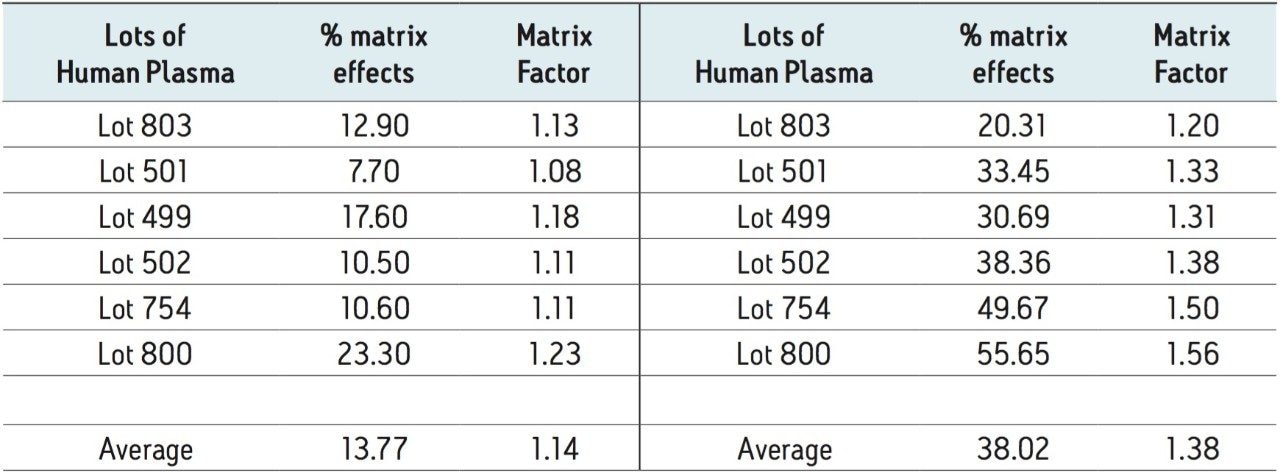
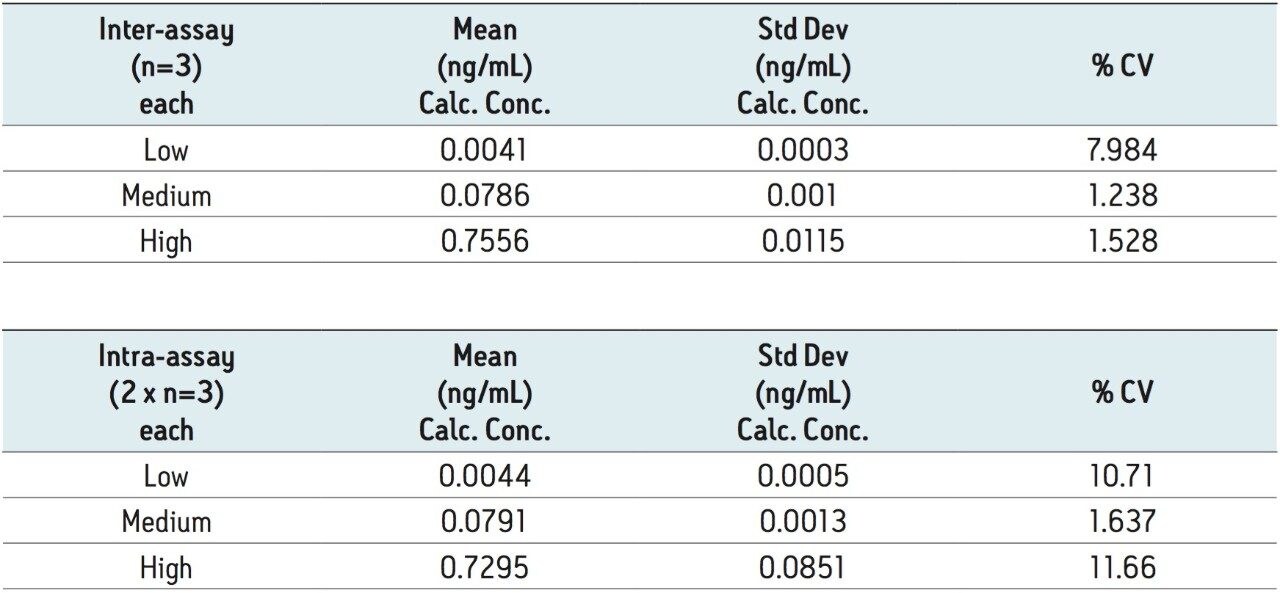
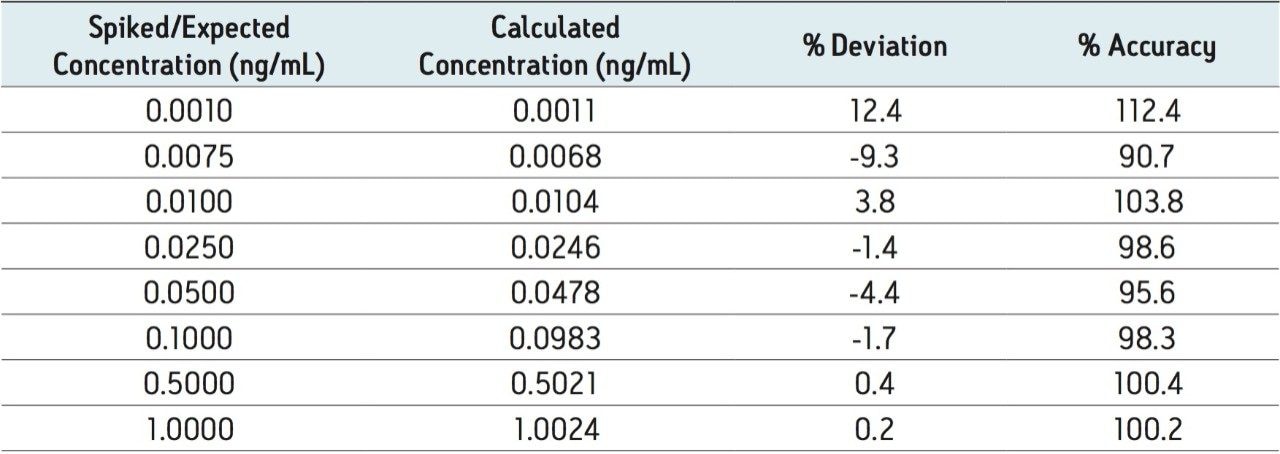
Ethinylestradiol (EE) is a common synthetic estrogen used in birth control formulations, and is often combined with other semi-synthetic estrogens. Estrogens are involved in development and maintenance of the female phenotype, germ cell maturation, and pregnancy. Males also utilize estrogen-based steroids for growth processes, nervous system maturation, bone metabolism and remodeling, and endothelial responsiveness. [1] Due to the presence of estrogen and related steroids in biological matrices, full chromatographic resolution of EE from these endogenous constituents is challenging. To accurately quantify EE, these endogenous compounds need to be efficiently removed, reduced, or separated utilizing a combination of sample preparation and chromatography. All remaining interferences after sample preparation must also be chromatographically resolved from the estrogen-based active pharmaceutical ingredient (API).
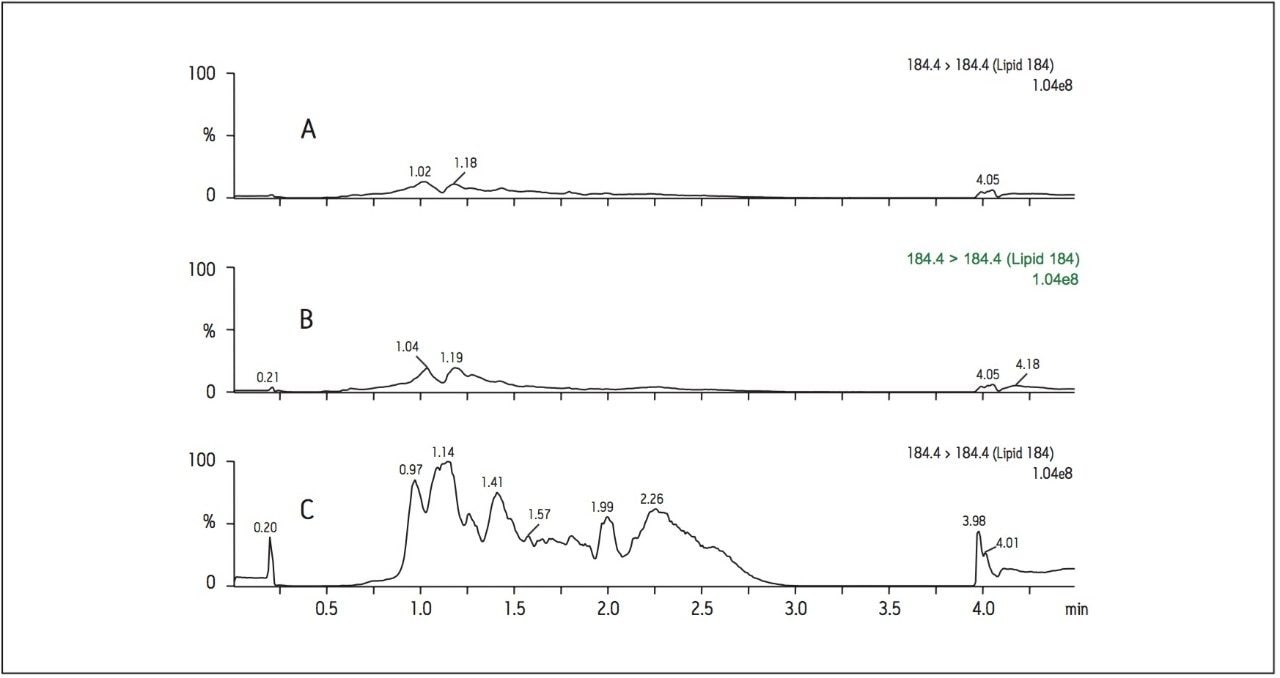
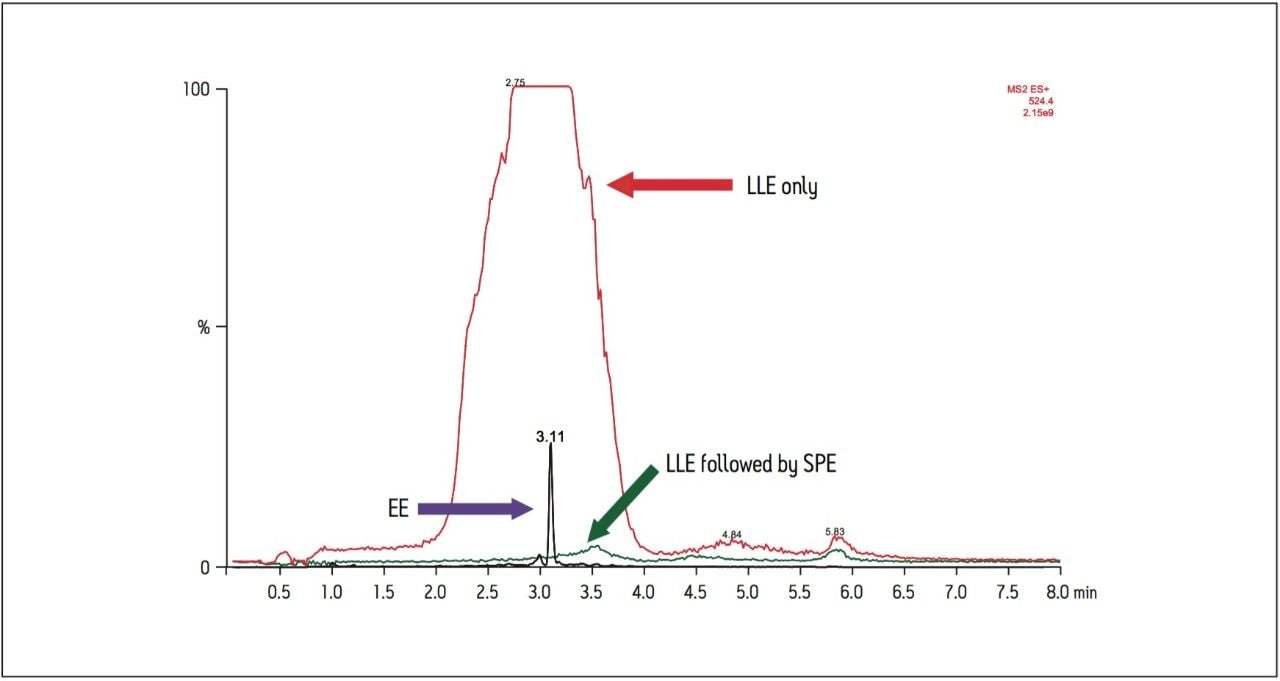
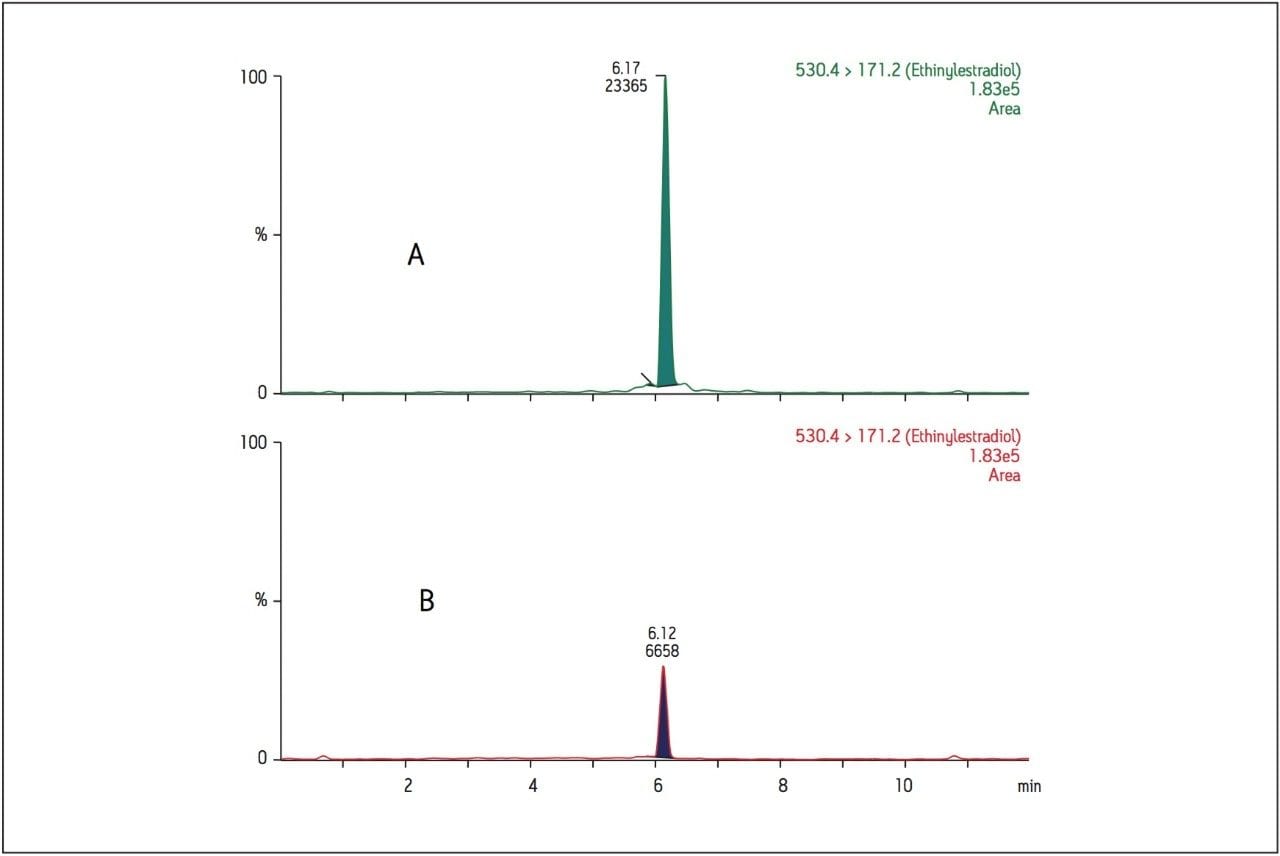
Recent published methods for EE utilize sample preparation consisting of liquidliquid extraction and derivatization. [1,2,3,4,5,6,7] These methods are capable of reaching detection limits of 0.01 ng/mL (10 pg/mL) in human plasma, a level that is no longer sufficient for low dose contraceptives. Current MS systems, although more sensitive for analyte detection, are also more sensitive to background, contamination, and overall cleanliness of samples and solvents. This makes routine achievement of even previously reported detection limits heavily reliant on additional sample preparation. Sample preparation methods must not only perform a clean-up of endogenous plasma interferences, but also concentrate samples to meet challenging limits of detection. For example, current birth control formulations require methods capable of achieving detection limits in the single pg/mL range. For this application, a limit of detection (LOD) of 1 pg/mL was required. For such challenging assays, each aspect of the method must be carefully optimized, including sample preparation, chromatographic separation, and spectrometric detection. In this study, sensitive and selective methods for ethinylestradiol (see Figure 1 for chemical structures) in human plasma were developed and partially validated.