The incidence of food allergies in industrialized populations has been increasing over the past decade.3,4 Many different food types cause adverse health effects for people susceptible to allergic reactions, ranging from mild unpleasant symptoms to deadly anaphylactic shock. Regulations5 that help reduce the risk of cross-contamination during food production and cover product labelling have been put into place as safeguards.
The most prevalent types of foods that can cause allergic reactions include peanuts, tree nuts, wheat, soybean, cow’s milk, hen’s eggs, fish, and crustacean shellfish. In particular, peanut allergy is a major problem due to the ubiquity of peanut use and severity of reactions.
The only effective way to prevent allergic reactions is to avoid allergen-containing food products. For those suffering with allergic reactions to certain food types, avoidance is very difficult unless the labelling on the packaging is clear, and no cross-contamination has occurred during the production process.
There are a variety of techniques available that analyze the presence of allergenic ingredients in food, the more popular techniques being enzyme-linked immunosorbent assay (ELISA) and DNA analysis. These methods often do not target a specific allergenic protein, but use a marker that is indicative of the offending food (e.g., trace of peanuts),6 and separate analytical methods are employed to target each allergen of interest.
More recently, the use of mass spectrometry (MS) for the detection of allergens has received increasing interest; in part due to its ability to have one platform to analyze multiple allergenic markers, as well as its ability to target the specific protein in its natural and modified state, plus the sensitivity of detection; trace levels are detectable with and without processing.
The use of liquid chromatography (LC) with MS combines the separation and identification of individual proteins to provide an unambiguous identification of allergenic proteins present in food products.
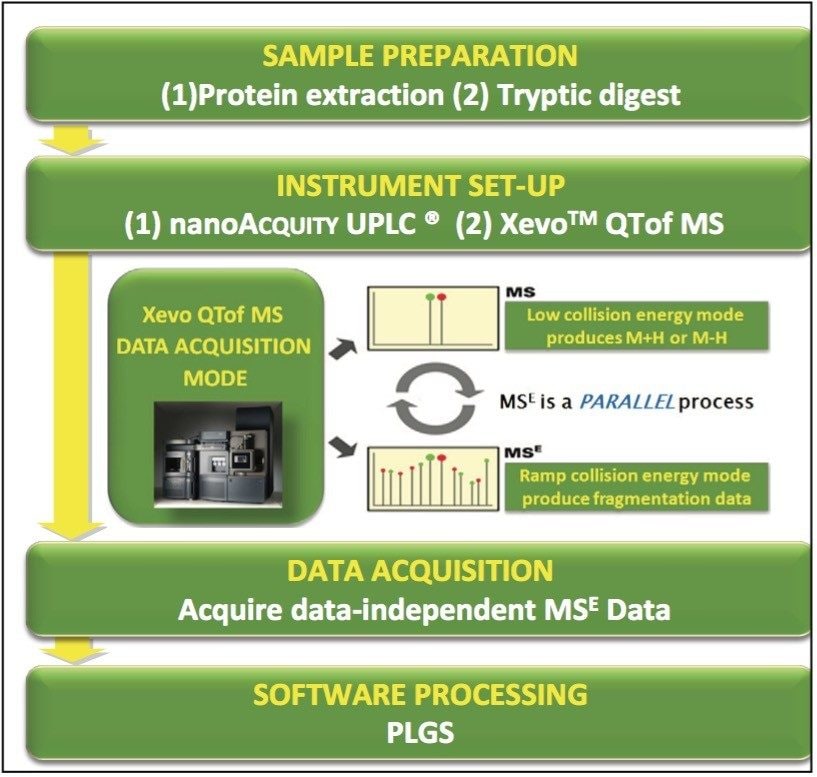
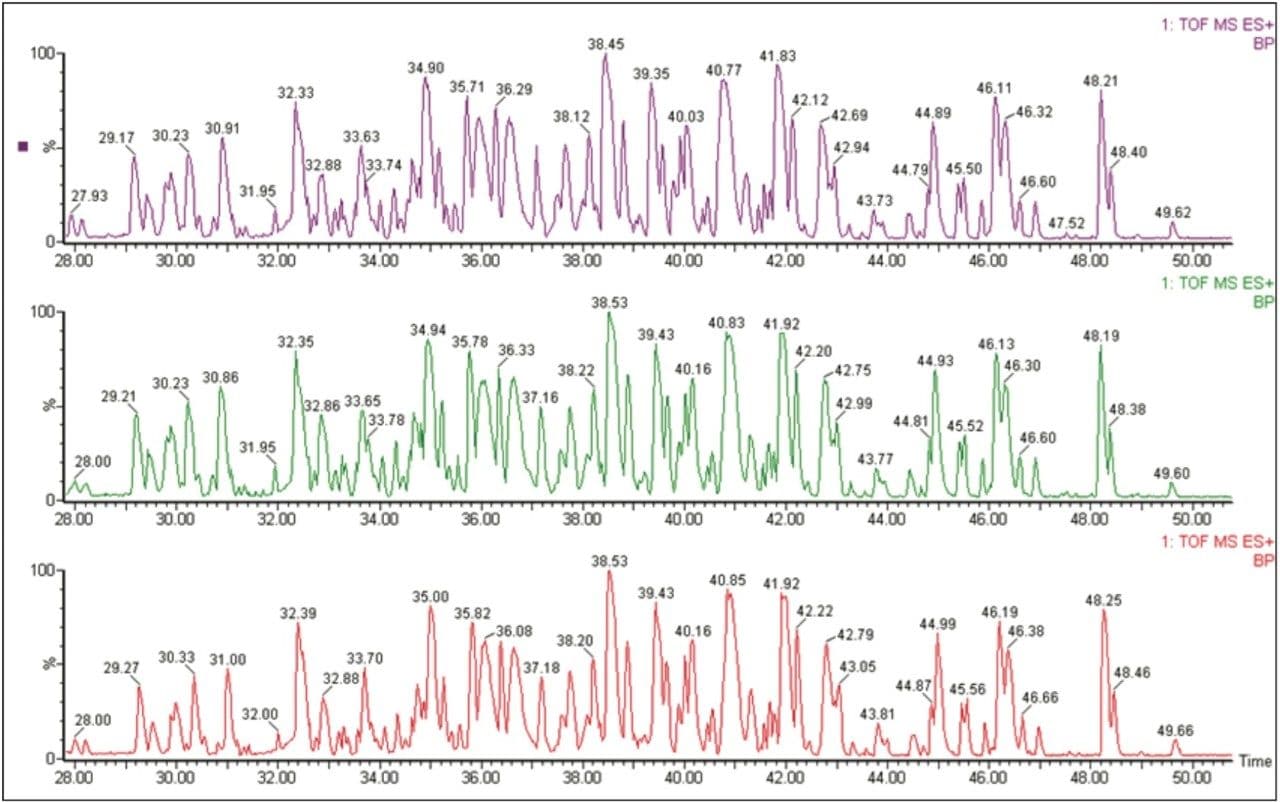
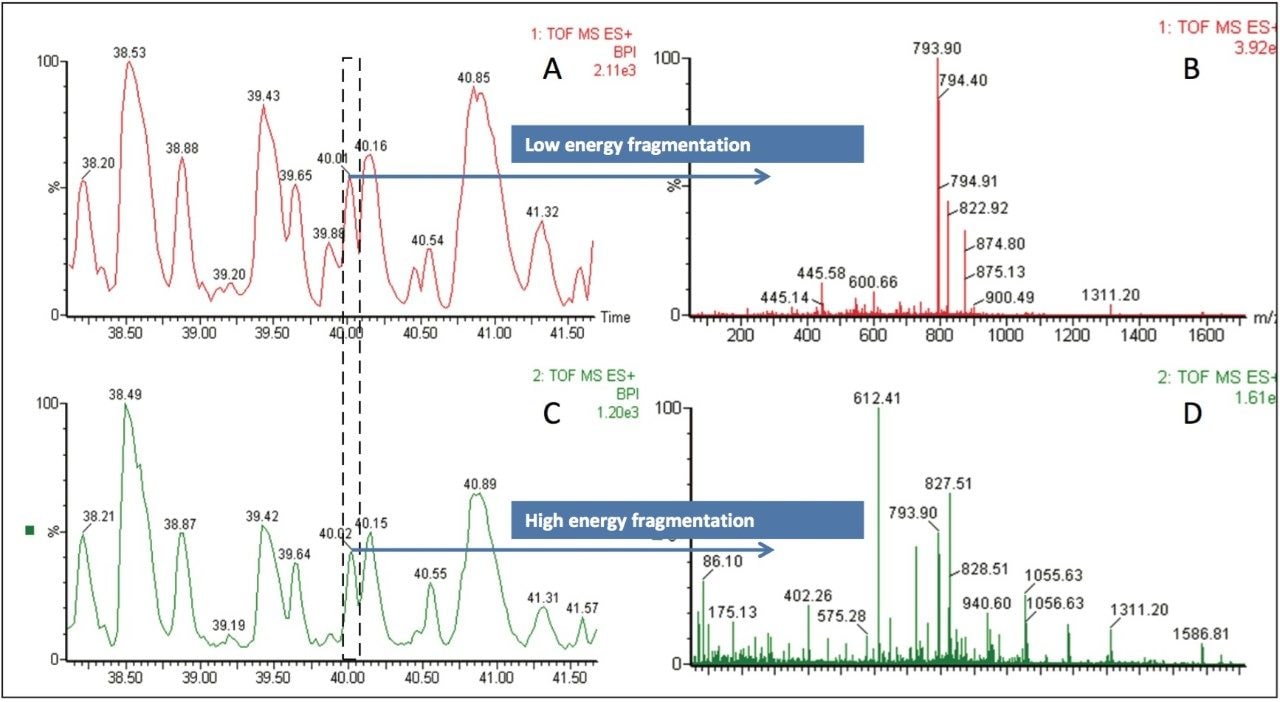
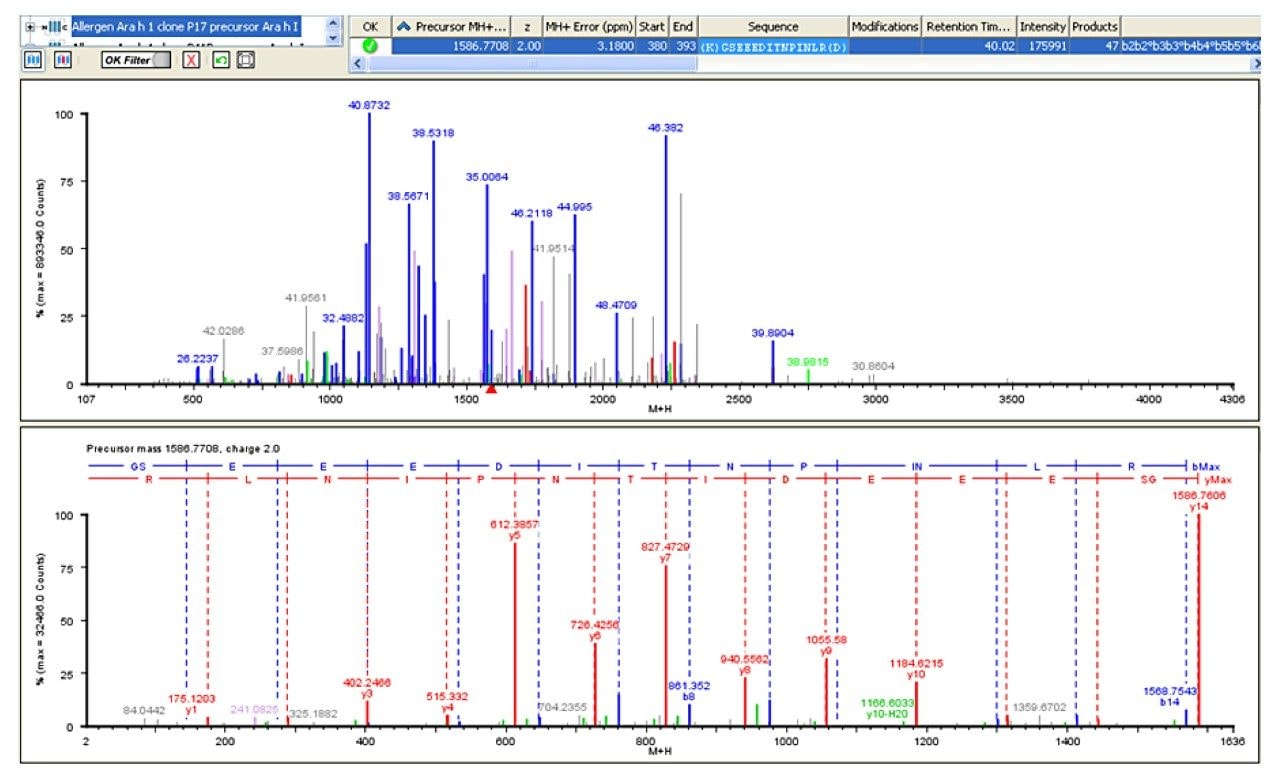
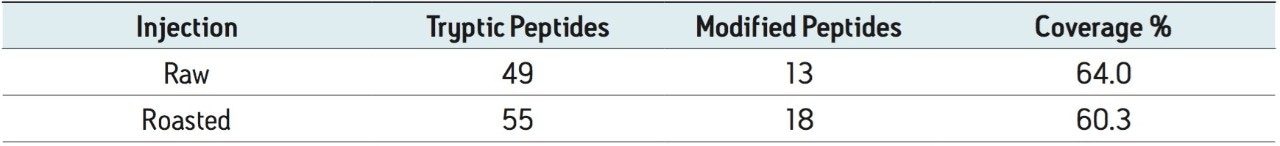
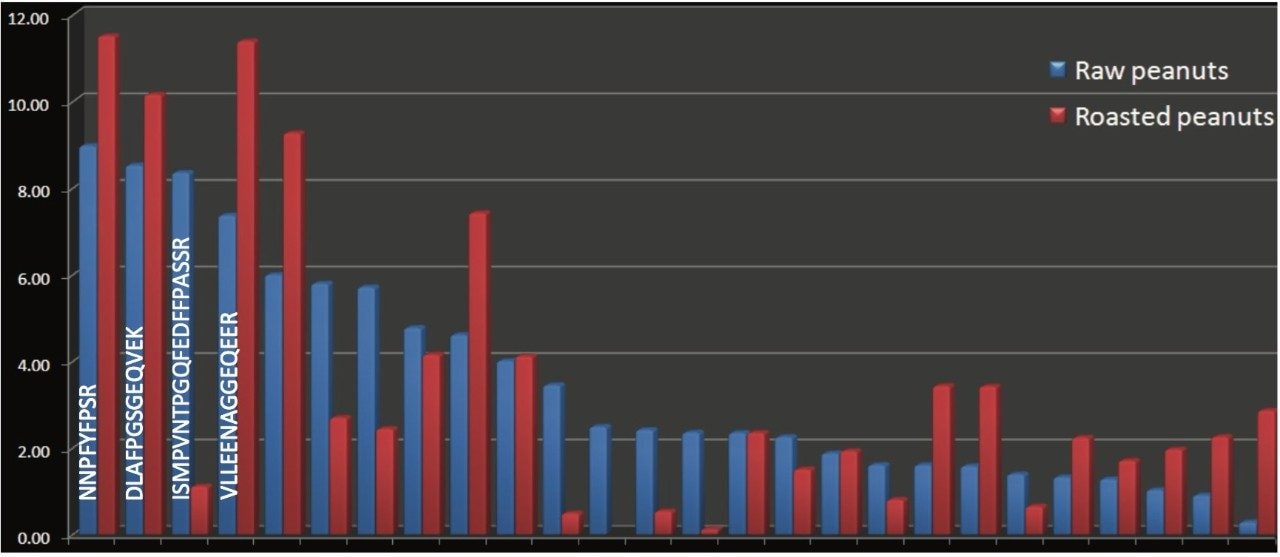
This work investigates the use of nanoACQUITY UPLC and Xevo QTof MS, and a food-proteomics approach to identify markers associated with the allergic reaction observed with peanuts, and to see if the markers can be observed in roasted peanuts. The technological processes used in the preparation of food products further contribute to the complexity of this system by inducing such phenomena as proteolysis and non-enzymatic glycosylation,7 and therefore there is a need to also assess the potential modifications that may be present.