Intact protein LC-MS analysis of a biotherapeutic provides a holistic view with a much simpler set of data than “divide and conquer” methods such as peptide mapping. The major tradeoffs for intact protein MS analysis are that the types of detectable modifications become more limited as protein mass increases and that modification sites cannot be deduced from the intact mass alone.
Assessing clone-to-clone or batch-to-batch variation for glycosylated therapeutic proteins is a common and routine task that often requires significant resources for manual data processing and results integration. When high throughput or rapid sample turnaround is desired, analytical approaches that provide rapid global information about a molecule are preferred over slower approaches that generate more detailed information.
For modifications such as oxidized methionines, there is roughly a 16 Da increase over the unmodified protein mass. These modifications can be readily detected at lower levels in the TOF mass spectra of smaller proteins (30 kD and below, e.g. antibody light chains), but require significant stoichiometry to be detected on a 50 kD protein (e.g. antibody heavy chains), and are not readily detectable on large proteins such as a 150 kD intact antibody. For this reason, intact antibody analysis is almost always used in combination with reduced antibody (LC/HC) analysis and peptide mapping studies for full characterizing batches of a therapeutic antibody.
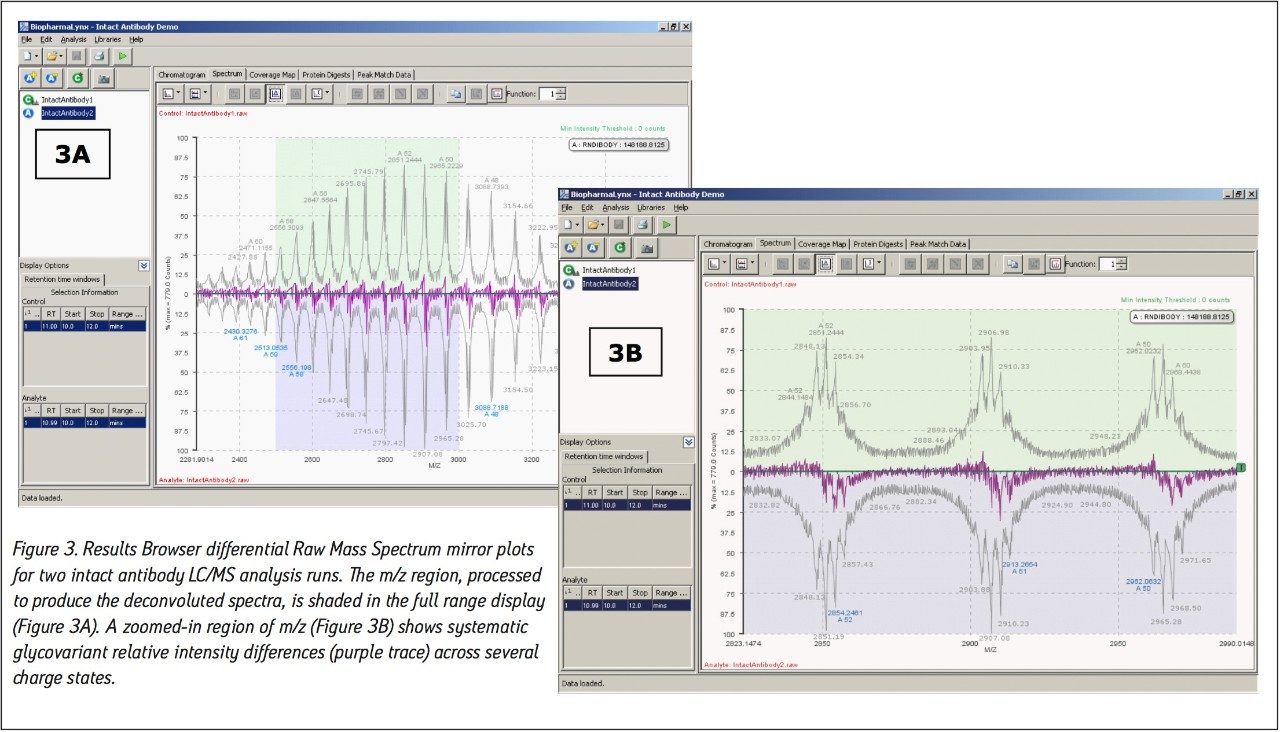
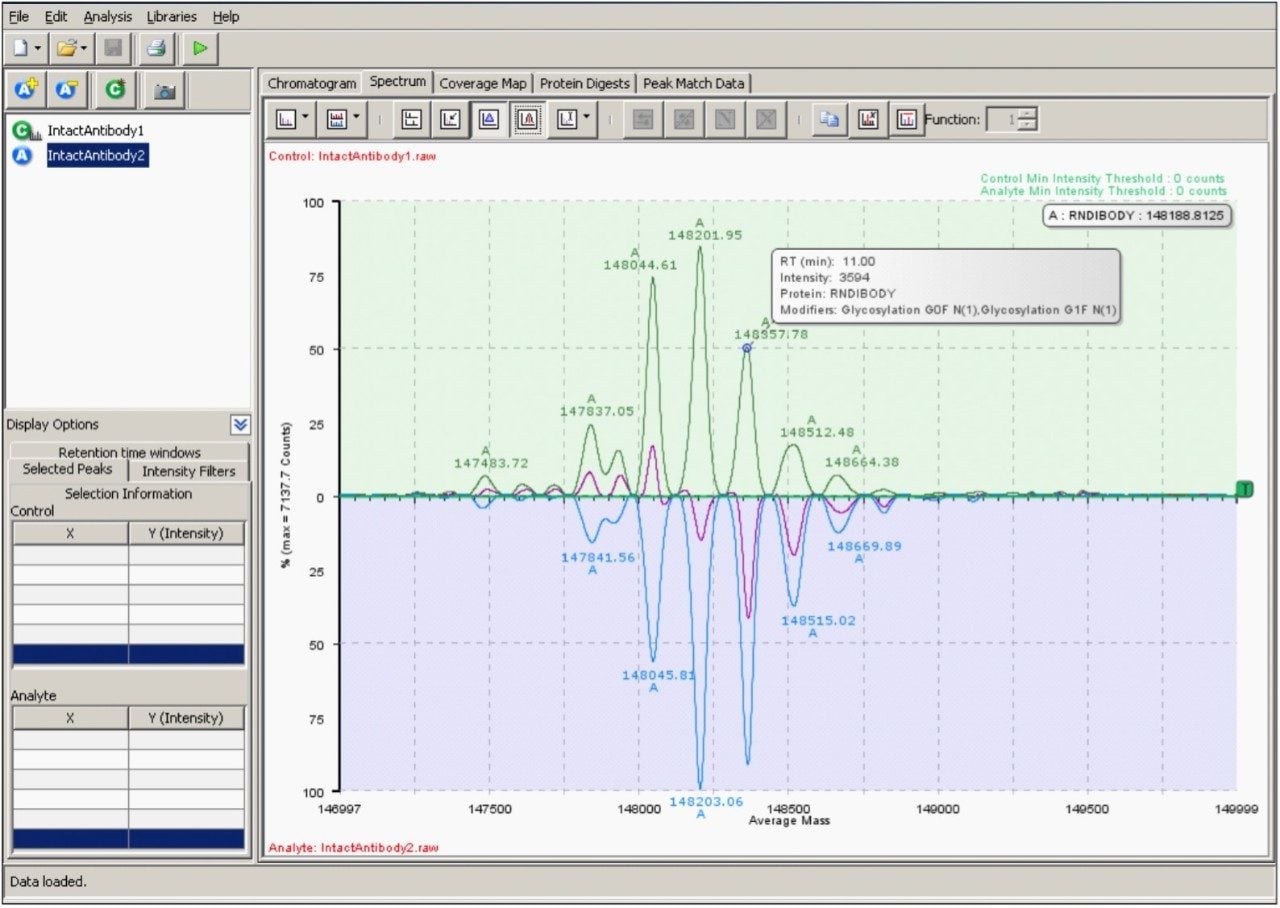
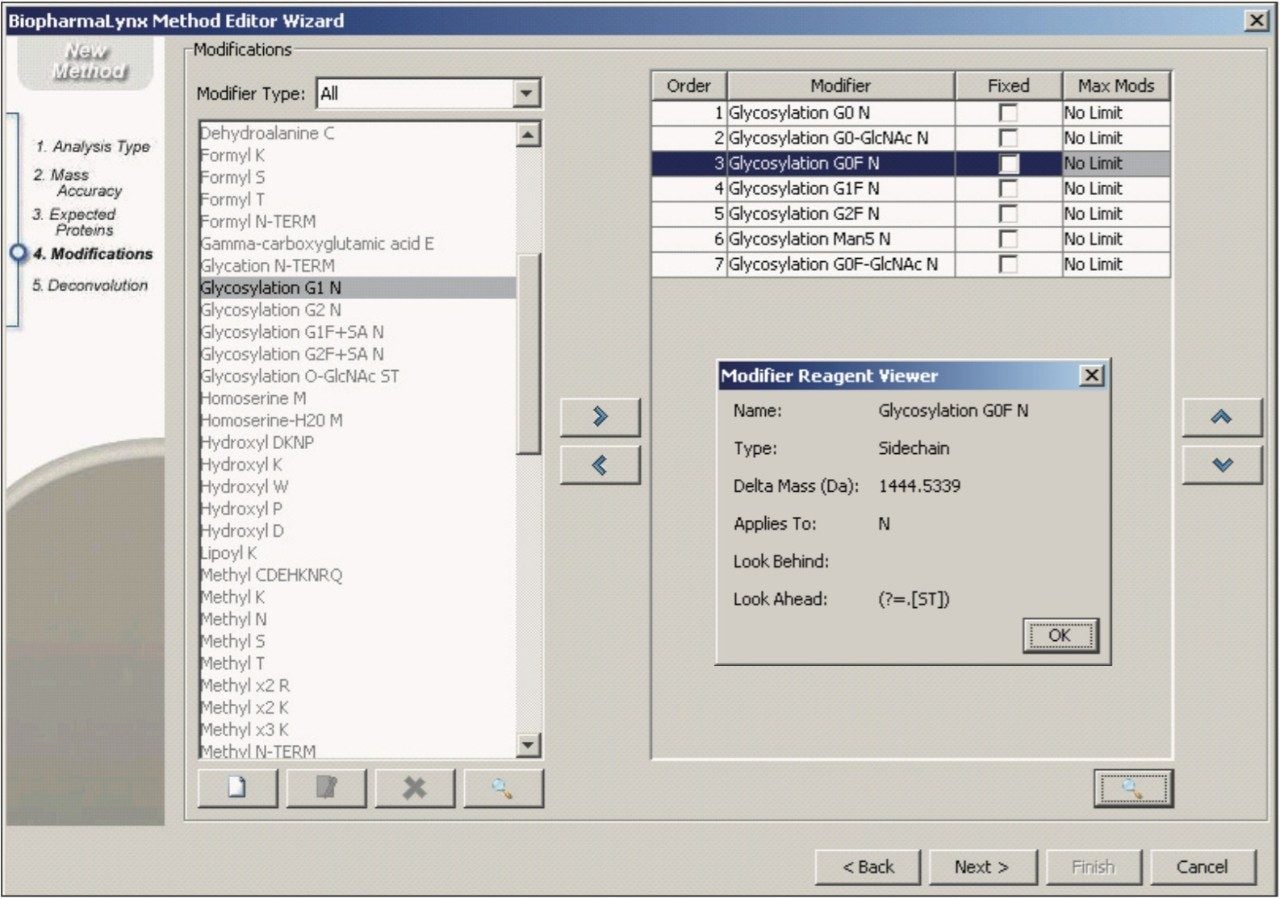
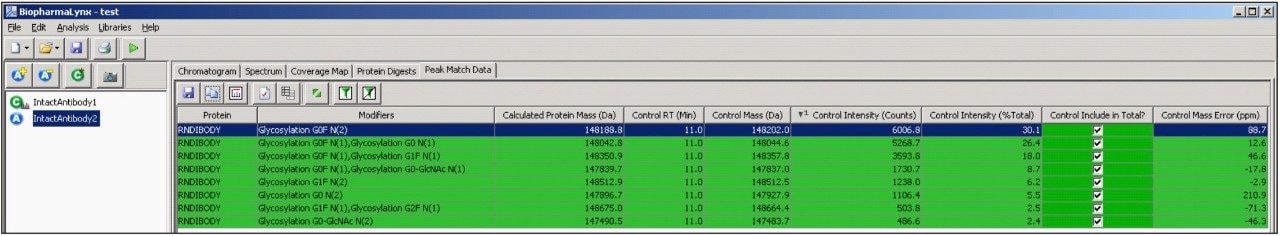
In general, there are two classes of protein variants that are typically scrutinized using mass analysis of an intact antibody: glycan structure heterogeneity where variation of core glycans on each heavy chain is extended by a series of 146 to 291 Da carbohydrate units and, the potentially inefficient proteolytic processing of heavy chain carboxy-terminal lysine residues (+128 Da).
Assessing this heterogeneity using intact antibody mass profiles can be useful for selecting a clonal expression cell line with desirable product attributes and monitoring the effects of process changes on a biotherapeutic. It is also a useful approach for demonstrating the consistency and comparability of individual batches of drug product.
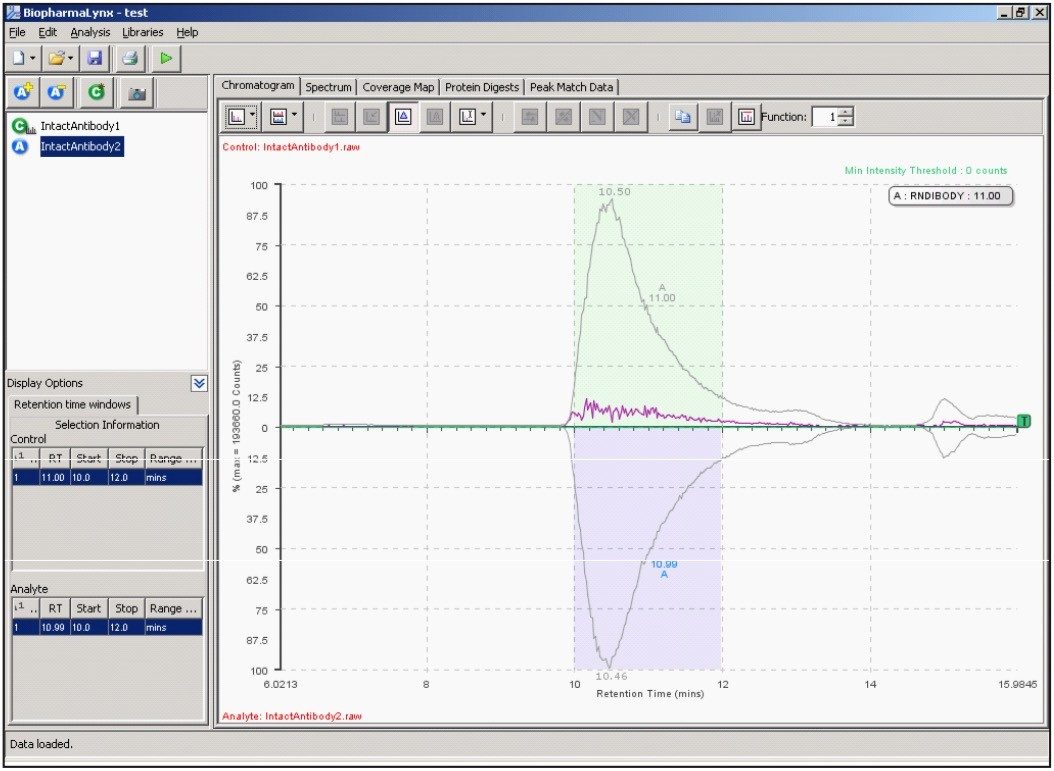
Comparative LC-MS analysis of intact antibodies can be accomplished using rapid LC-MS methods.1-3 Using proper methodology, this type of analysis is robust, and pharmaceutical companies have acquired intact mass data on literally thousands of proteins using OpenLynx enabled open access LC-MS analysis stations.4-6 The resulting deconvoluted intact mass information found in an OpenLynx report is usually sufficient for routine mass confirmation of a recombinant protein, but often proves limiting for in-depth characterization or comparative profiling studies of biotherapeutics.
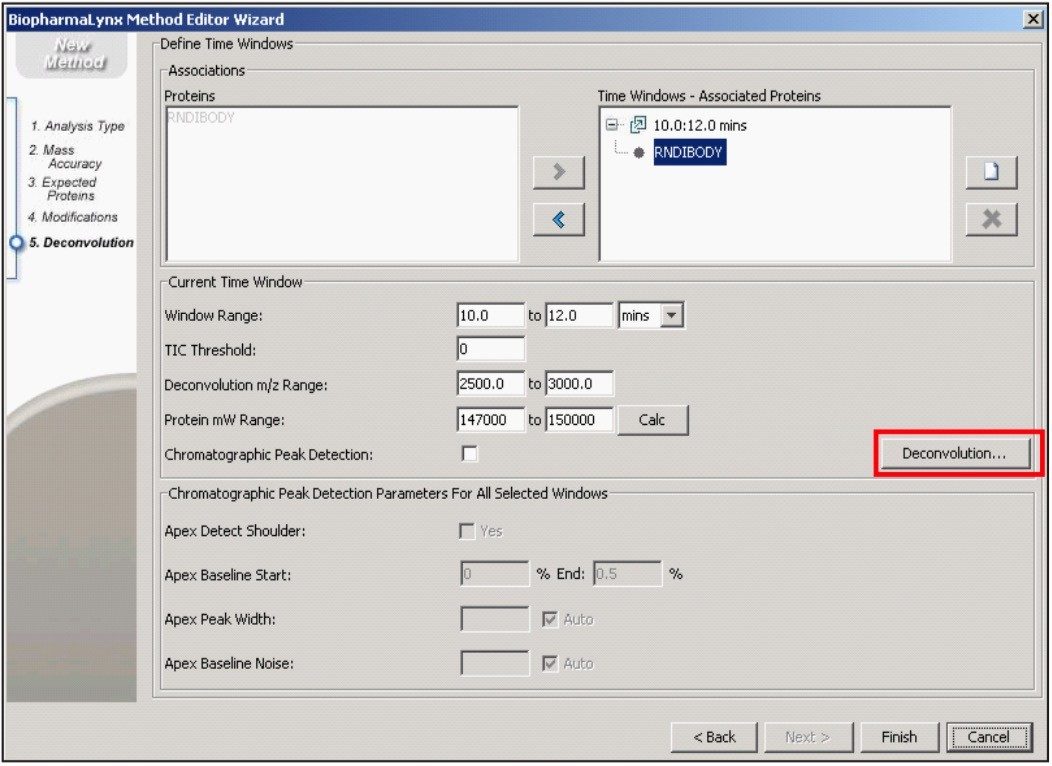
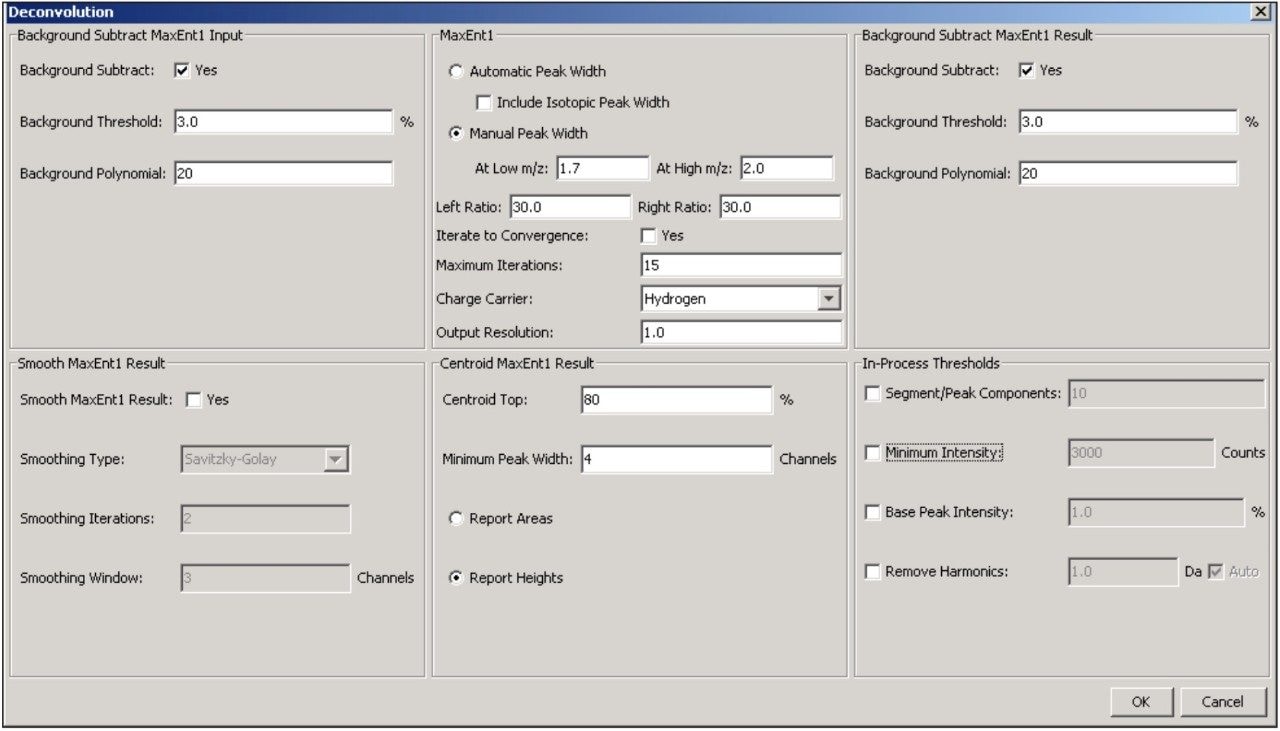
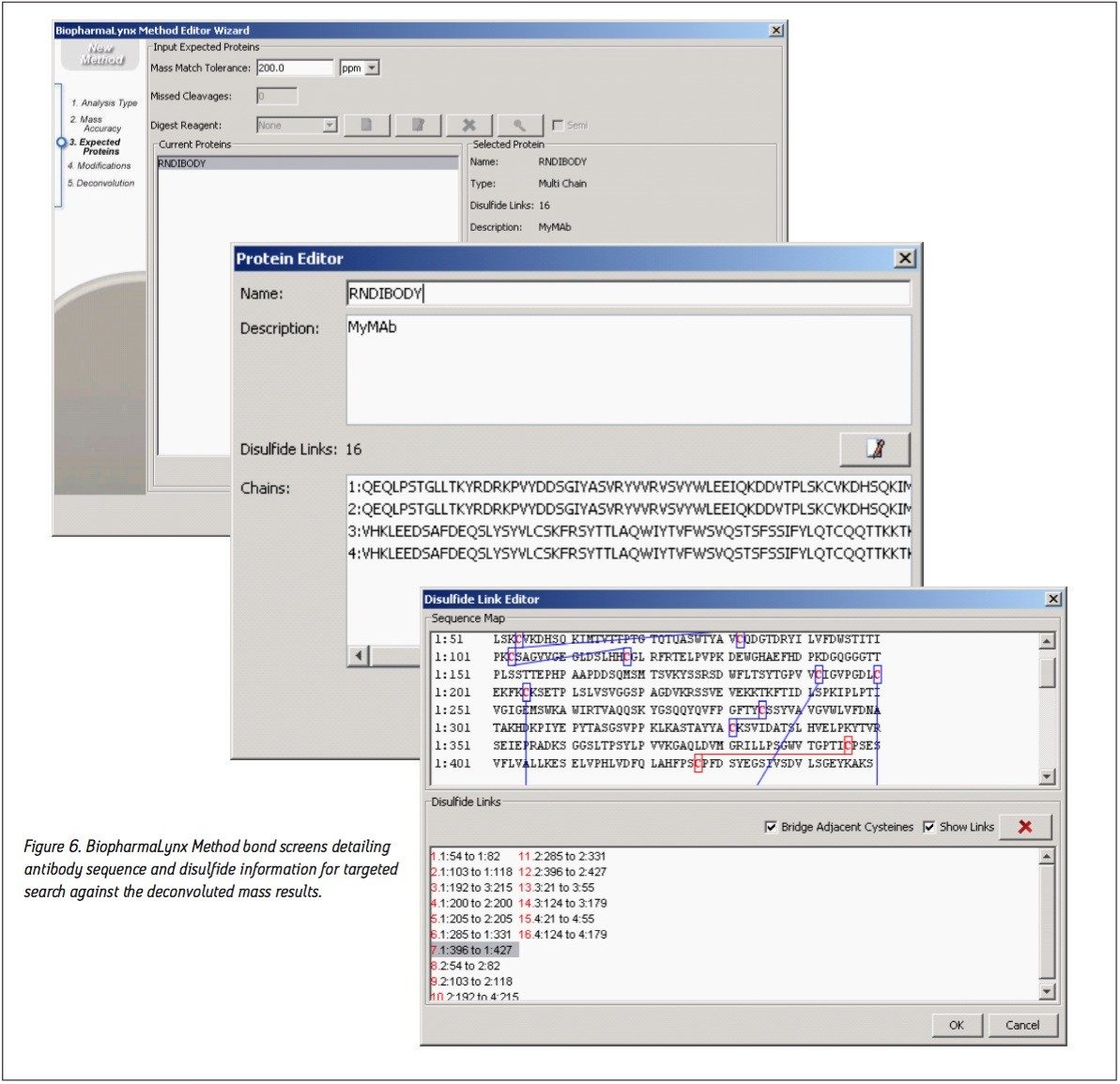
BiopharmaLynx intact protein analysis workflows are designed to expand functionality for automated batch processing of data by providing additional processing capabilities and related deconvoluted masses to targeted proteins and their variants.