Application 1: Identity test
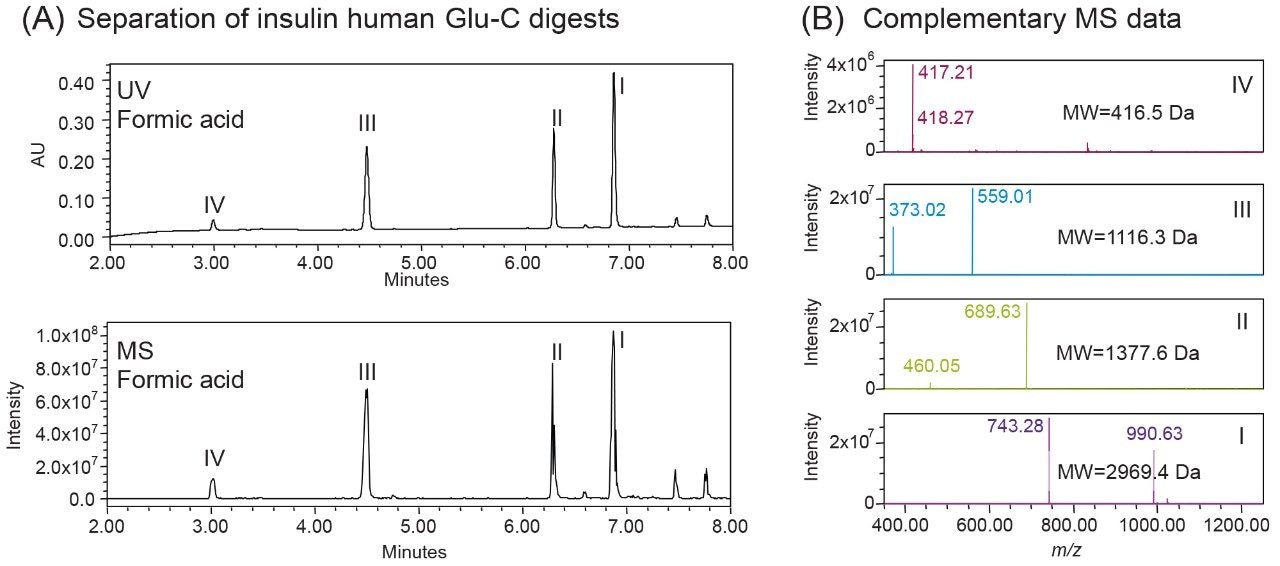
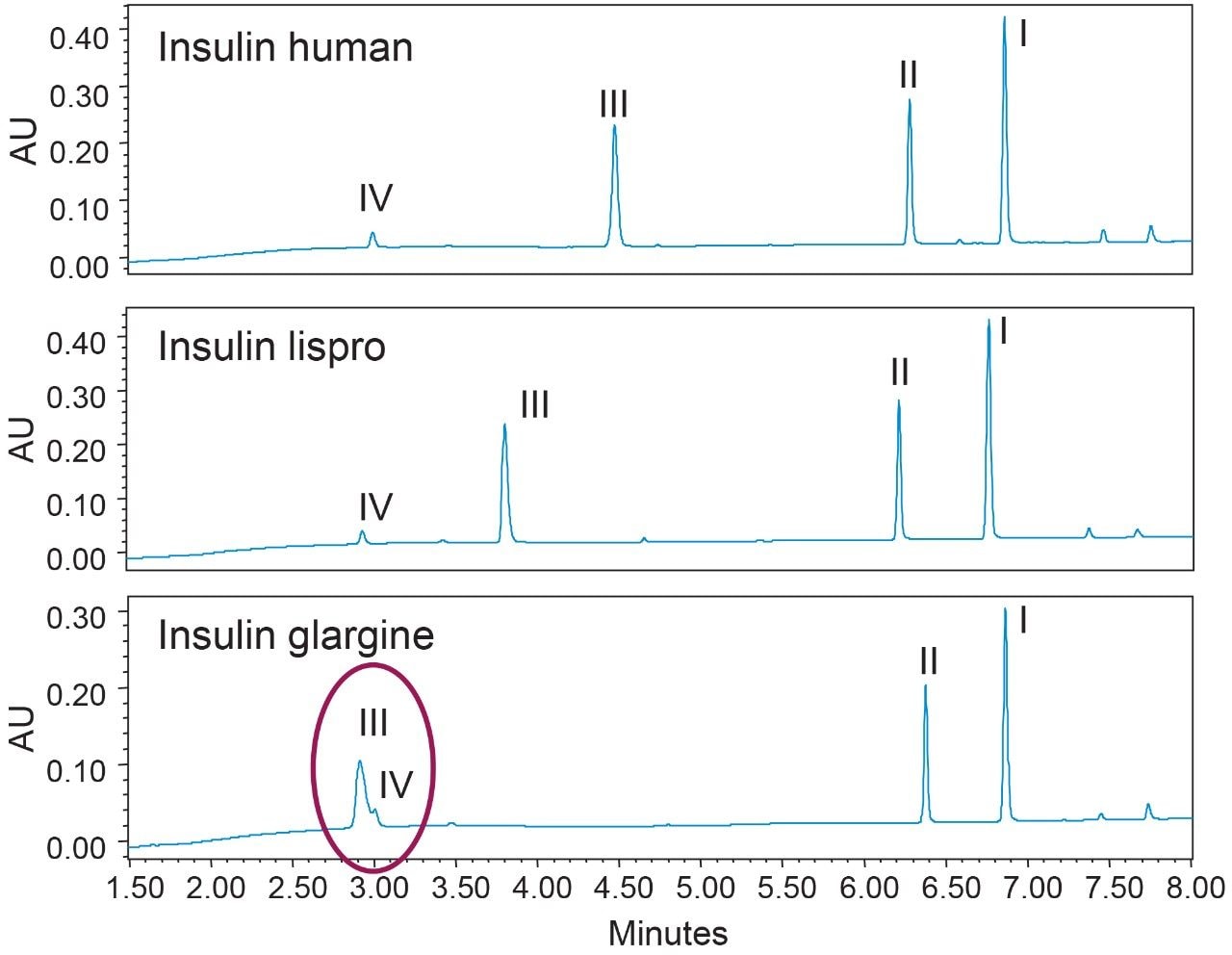
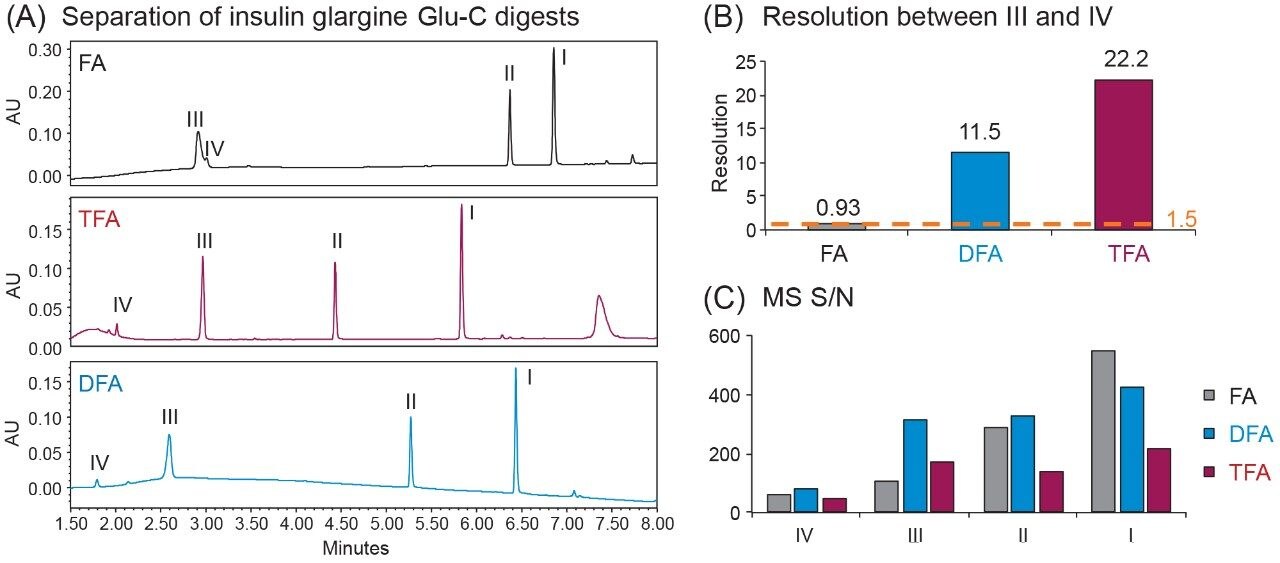
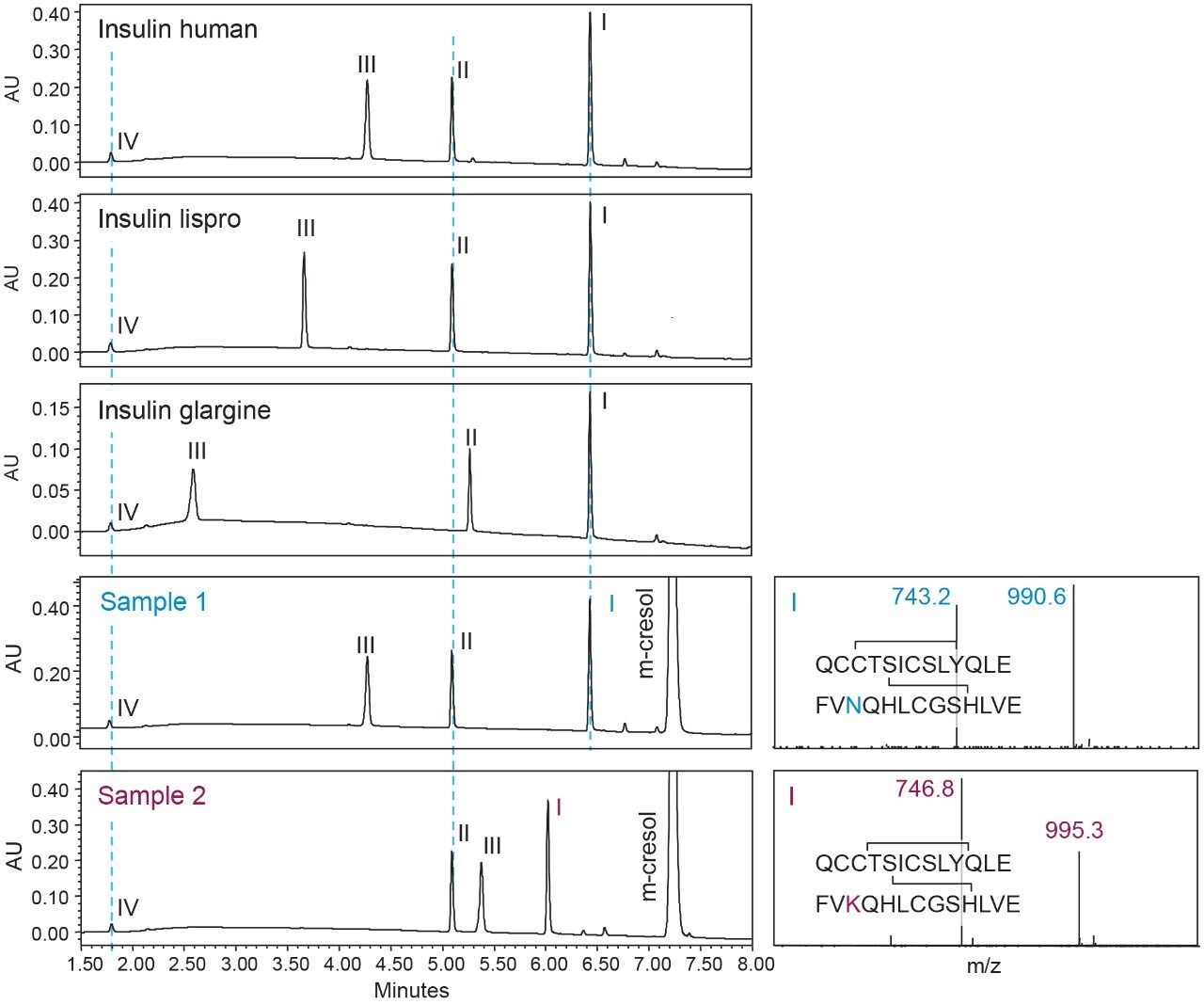
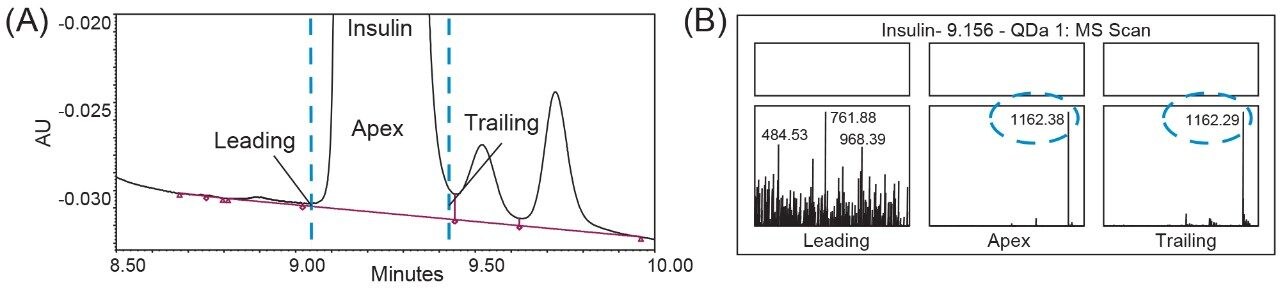
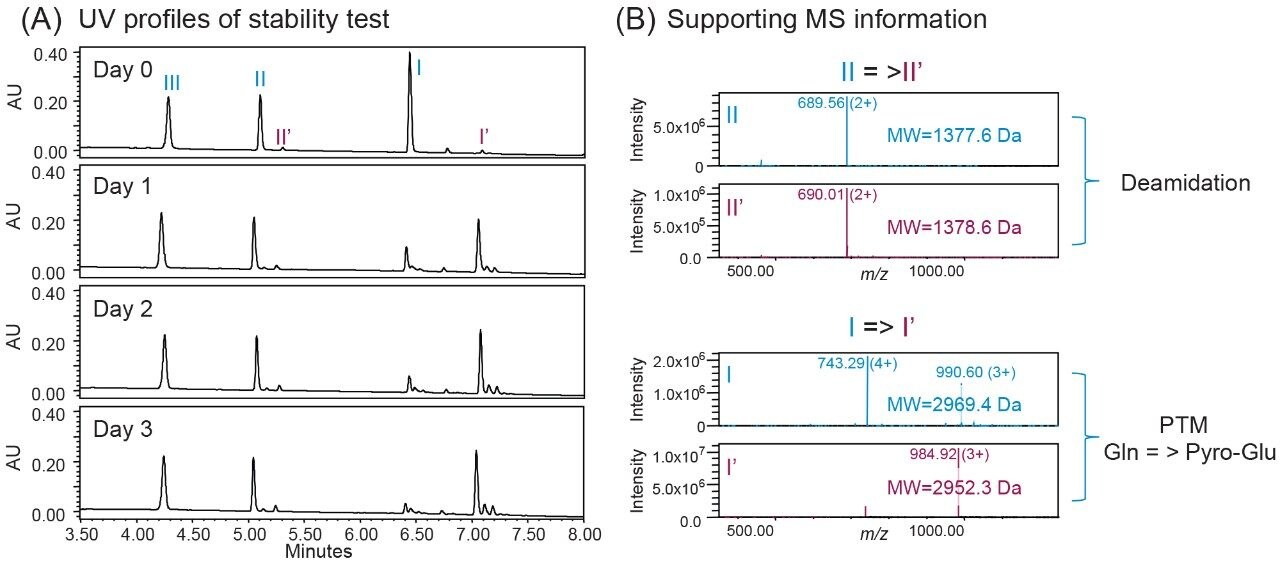
One of the benefits of adding MS detection to UV-based identity tests is the complementary mass information provided for investigation of the failed tests. Using the same gradient as the DFA in Figure 3, an LC-UV/MS based platform method was developed with three insulin analogues as reference standards, including insulin human, insulin lispro, and insulin glargine. As shown in Figure 4, all peaks were well resolved while the peptide fragments (Peak I, II, IV) with the same peptide molecular weight were aligned across samples as indicated by the blue dash lines. Using this platform method, an identity test was performed on two insulin drug products: Sample 1 and 2. As shown in Figure 4, the retention times of peptide peaks in sample 1 were aligned well with insulin human, while Peak I and III of Sample 2 shifted significantly. According to the collected MS data, a delta mass difference of 14.7 Da and 100.2 Da was observed for fragment I and III respectively (Figure 4), suggesting the inconsistency in retention times was caused by a single amino acid change in fragment III (KT to E) and I (N to K). Based on the sequence information, Sample 2 was identified as another insulin analogue: insulin glulisine. Collectively, the developed method demonstrates high robustness and specificity as a platform assay for identity test of various insulin analogues.