Intact mAb Mass Analysis
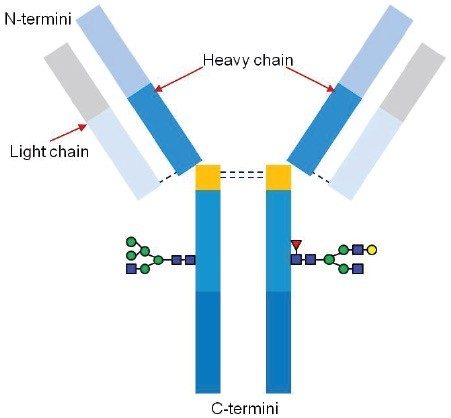
The LC-MS-based characterization of an intact mAb is often used alongside peptide mapping to profile modifications such as glycosylation, C-terminal truncation, glycation that can be mass resolved on a high resolution mass spectrometer (HRMS). In mAb characterization, intact mass measurements are often challenged by the extent of user intervention required in data analysis. UNIFI completely automates the analysis by streamlining data acquisition, processing, and reporting into one integrated workflow for both intact mAb and subunits analyses. Data analysis is simplified for the accurate mass measurements, modification confirmation, and relative abundance quantification. In this application, we demonstrate the use of intact protein workflow in UNIFI to profile modifications of the NIST mAb reference material.
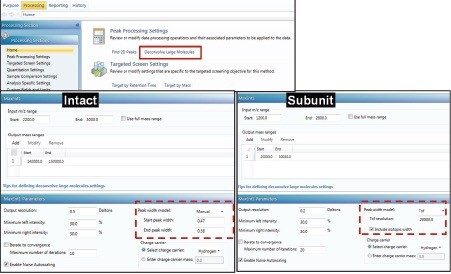
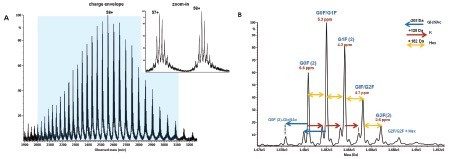
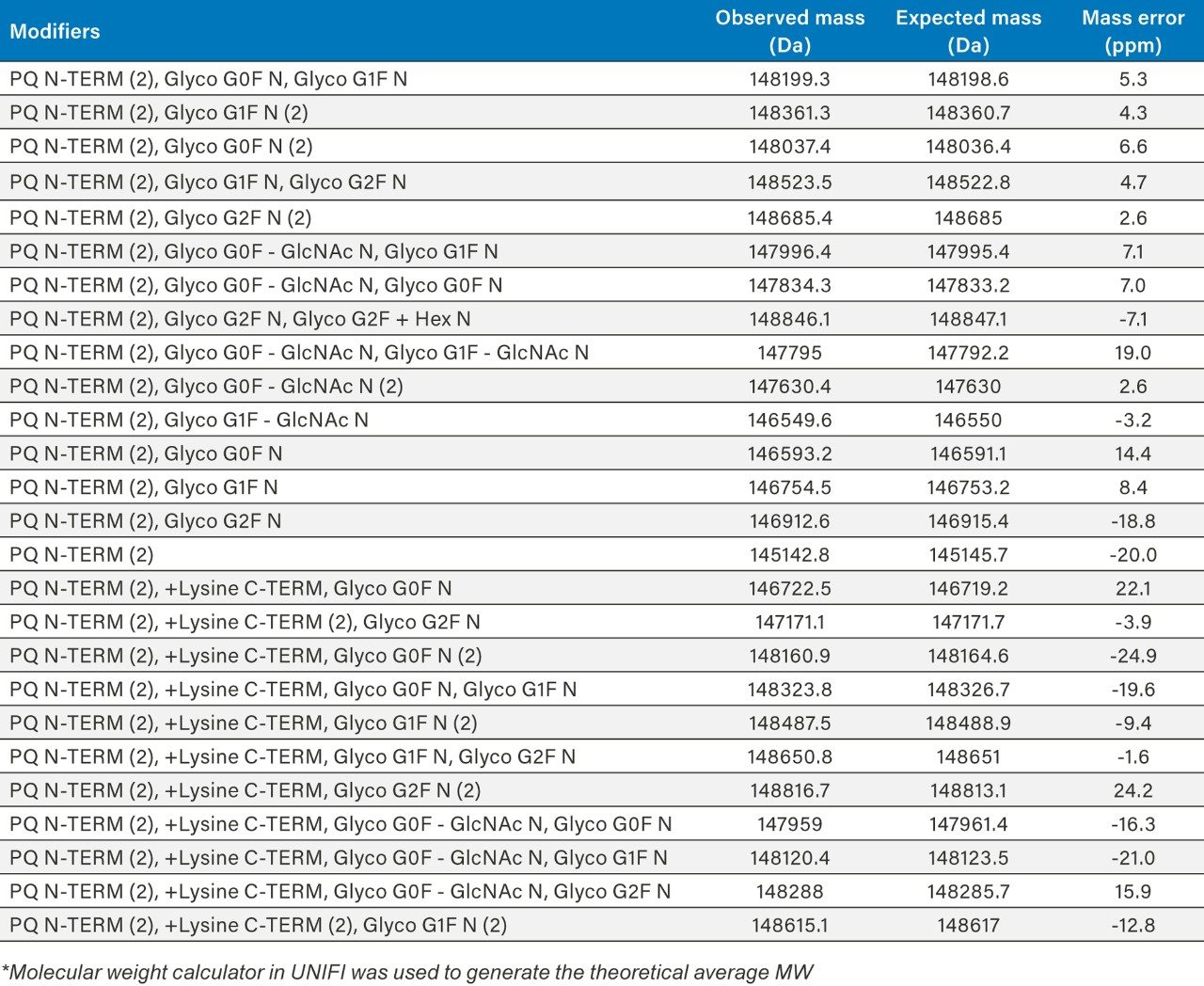
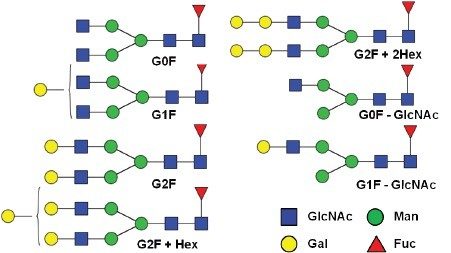
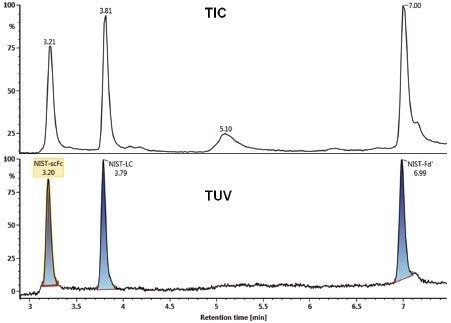
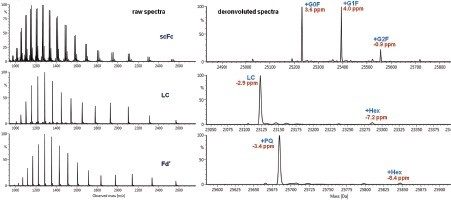
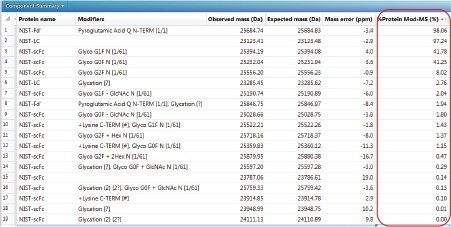
The summed raw spectrum (Figure 4A) presents the charge envelope for 0.1 µg of the intact NIST mAb (on-column) and the inset presents the zoomed-in region to show the peaks representing its five most abundant glycoform combinations. The UNIFI intact protein workflow has a MaxEnt1 charge deconvolution algorithm capable of generating the zero charge spectra using “manual” or “Tof” peak width models for the m/z range of interest. Here, the manual peak width model with user-defined start and end peak widths (full width at half maximum/ FWHM) was used. For an example, the selected peak widths of 0.47 and 0.58 (Figure 3) represent the respective FWHM for the most abundant species at the low and high ends of the charge envelope (m/z 2100–3000 mass window) as given in Figure 4A. The charge deconvoluted intact NIST mAb data and peak assignments are illustrated in Figure 4B. The most abundant glycoforms are bi-antennary structures; however, glycoforms identified by UNIFI also consisted of low levels of galactose-alpha-1,3-galactose containing glycoforms (Table 1). The structures of all glycans were later confirmed and quantified using a released N-glycan assay (data not shown).
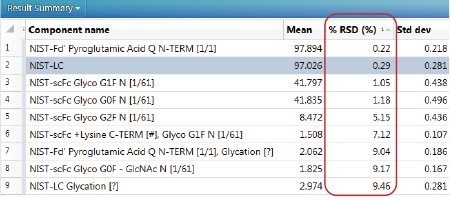
In addition to N-glycans, three other modifications: N-terminal pyroglutamic acid formation, C-terminal lysine truncation, and glycation were also investigated at the intact mAb level. The glycation analysis was performed after treating the mAb with PNGase F to remove all N-linked glycans. The MS response was used for the relative abundance measurements. In summary, we observed 10.5% of C-terminal lysine variants, which correlated very well with the peptide level measurement (data not shown), 14.6% of overall glycation (Table 2 and 3) and N-terminal pyroglutamic acid modification at >99% level. Our measurements are in good agreement with the published data.3