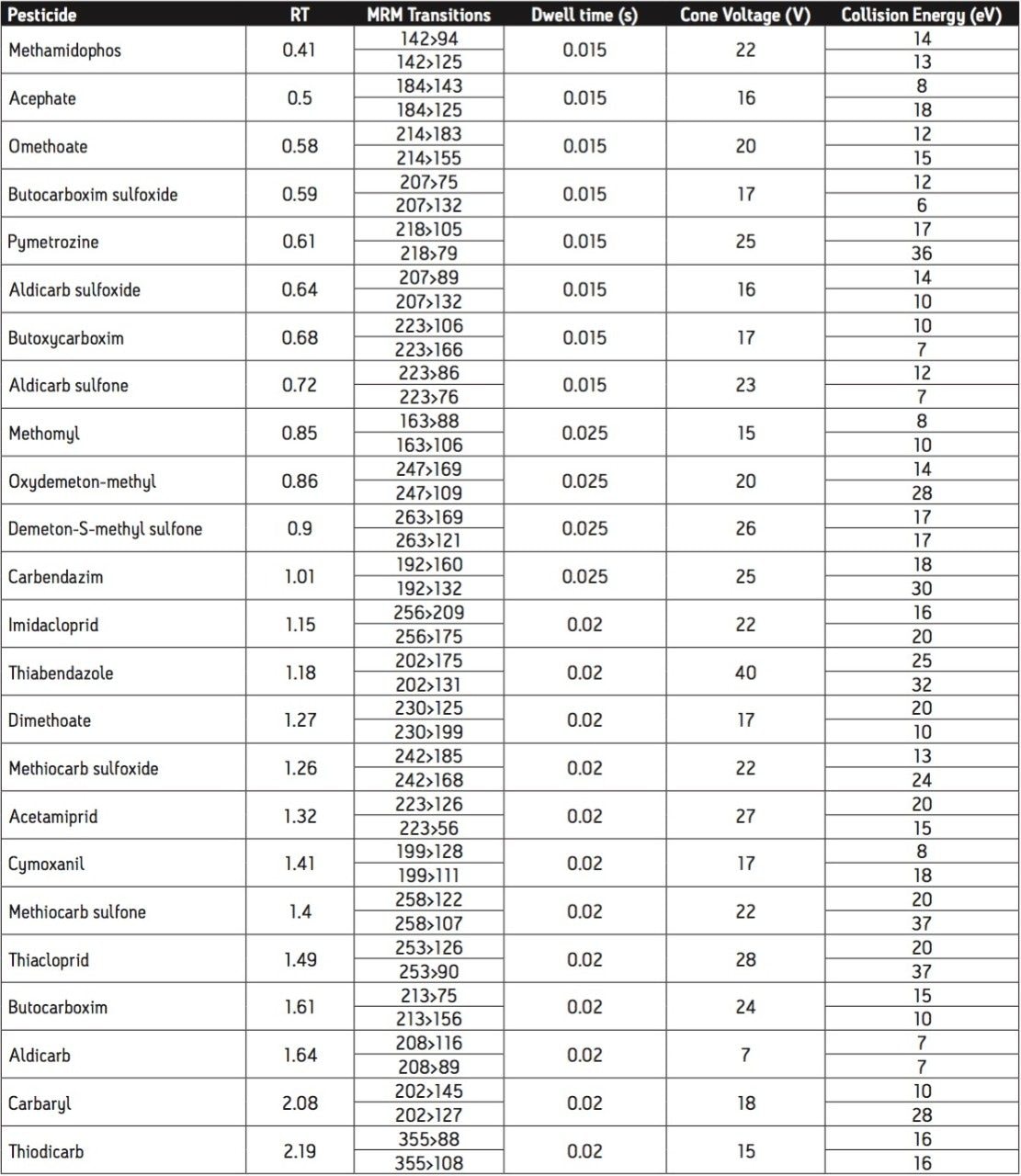
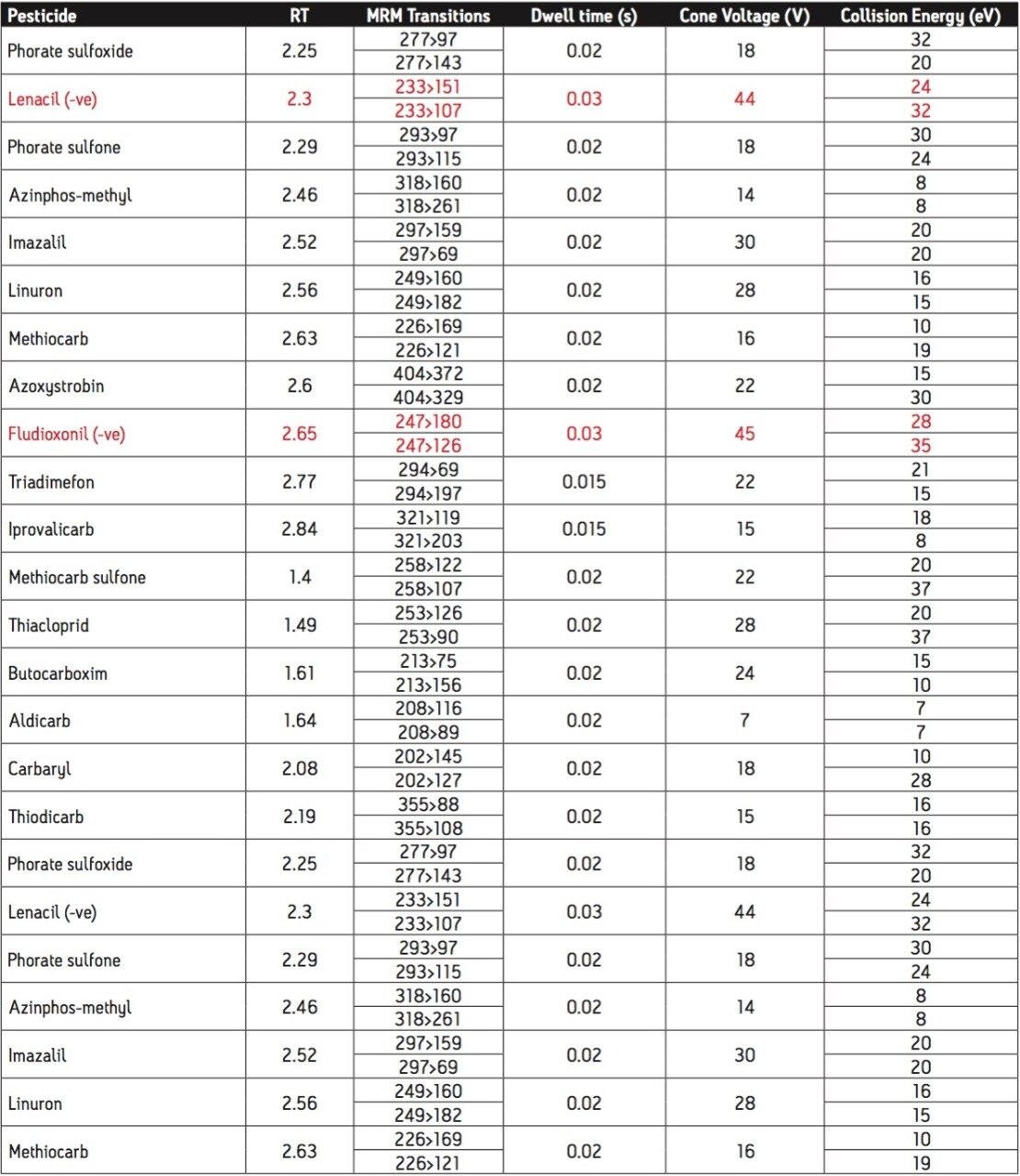
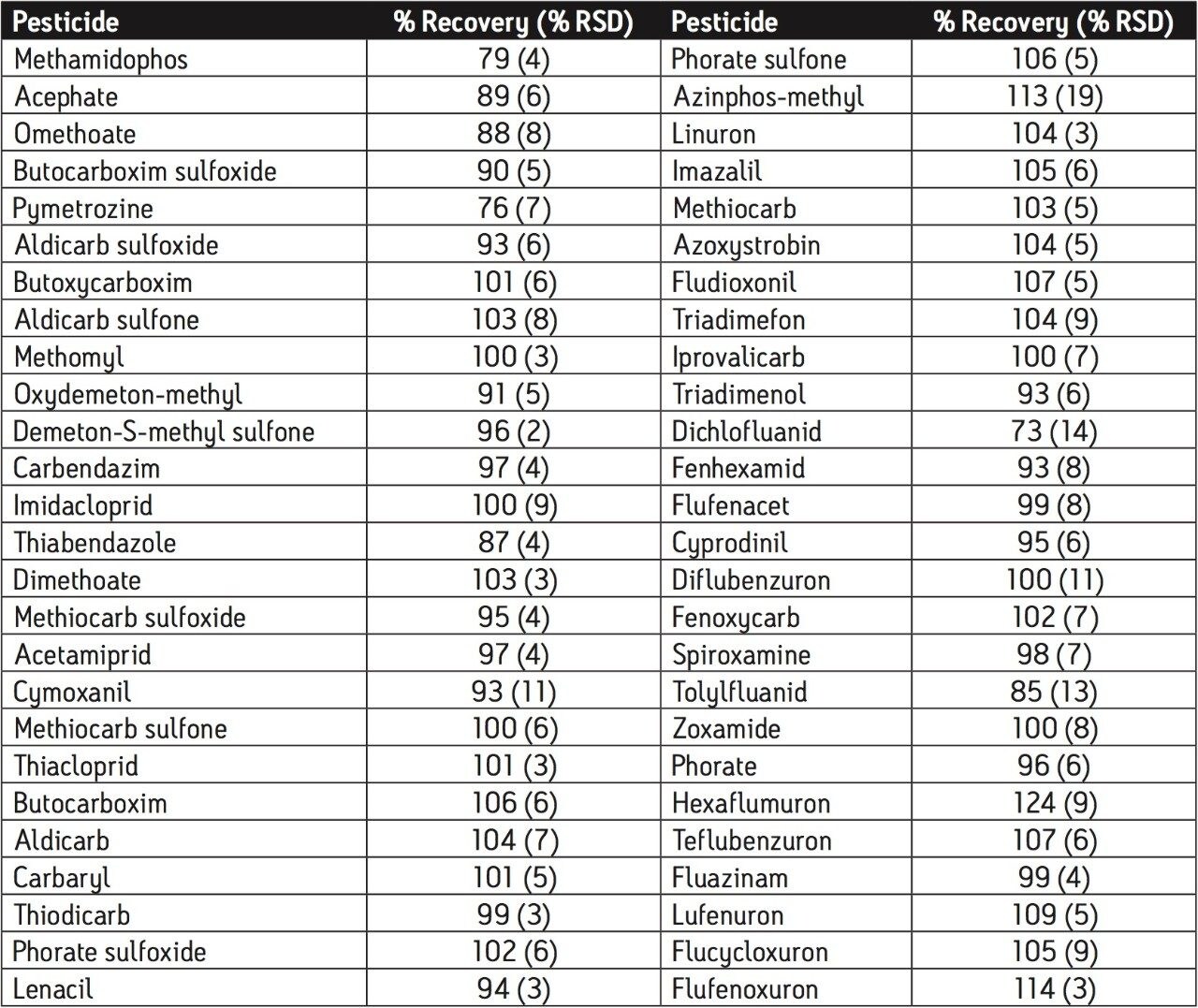
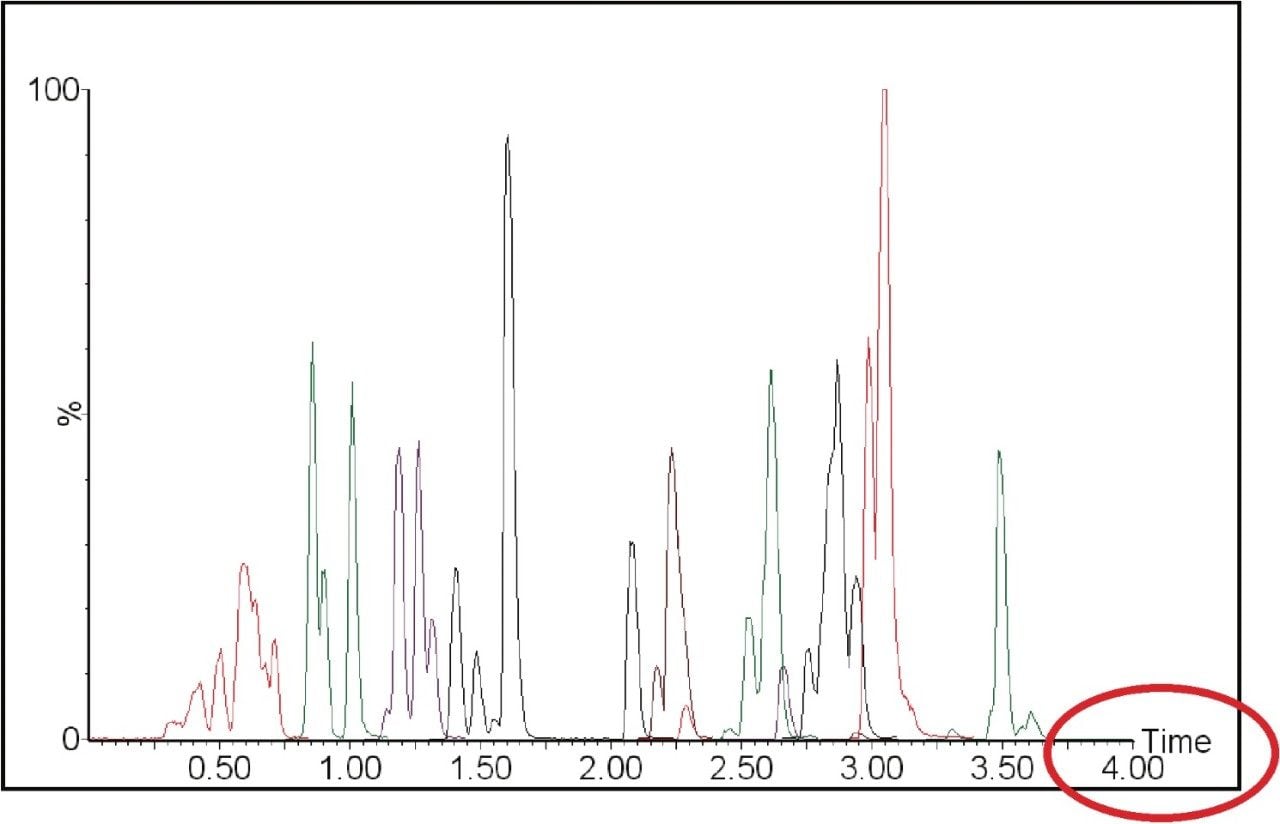
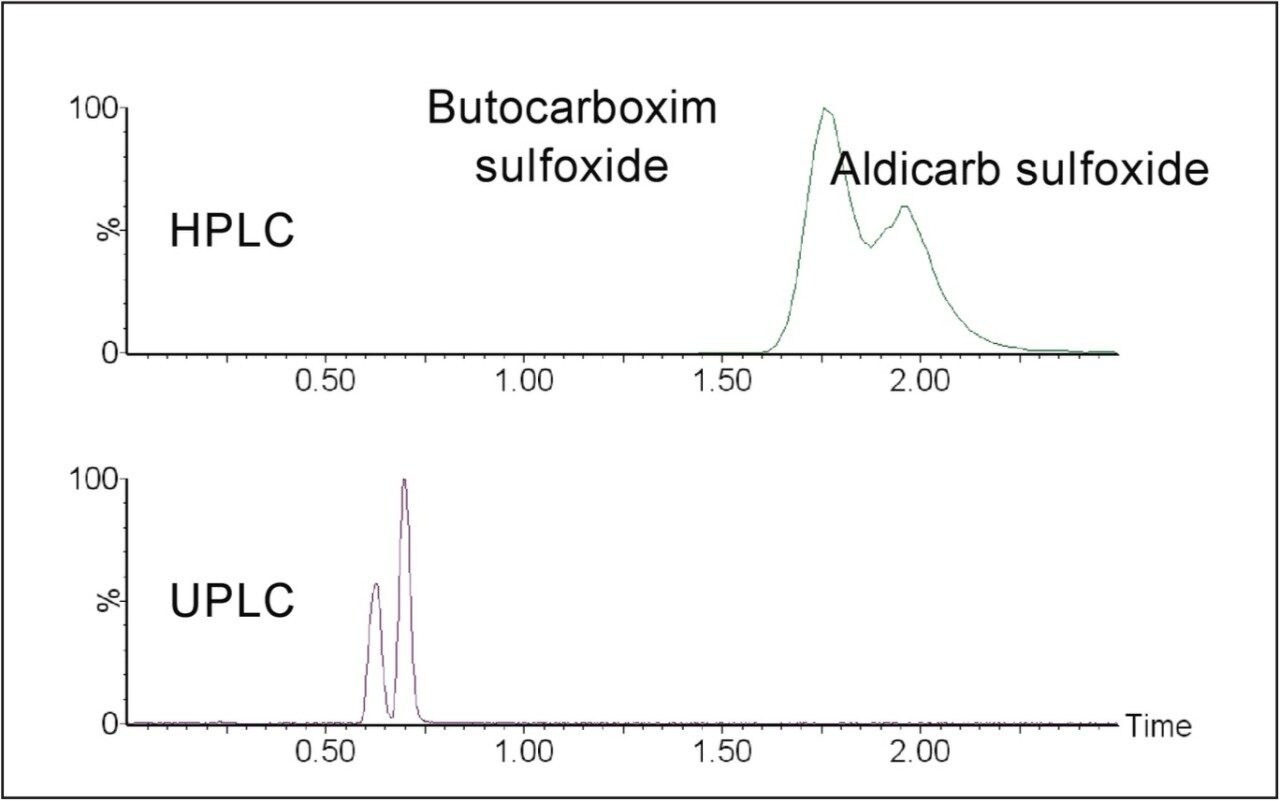
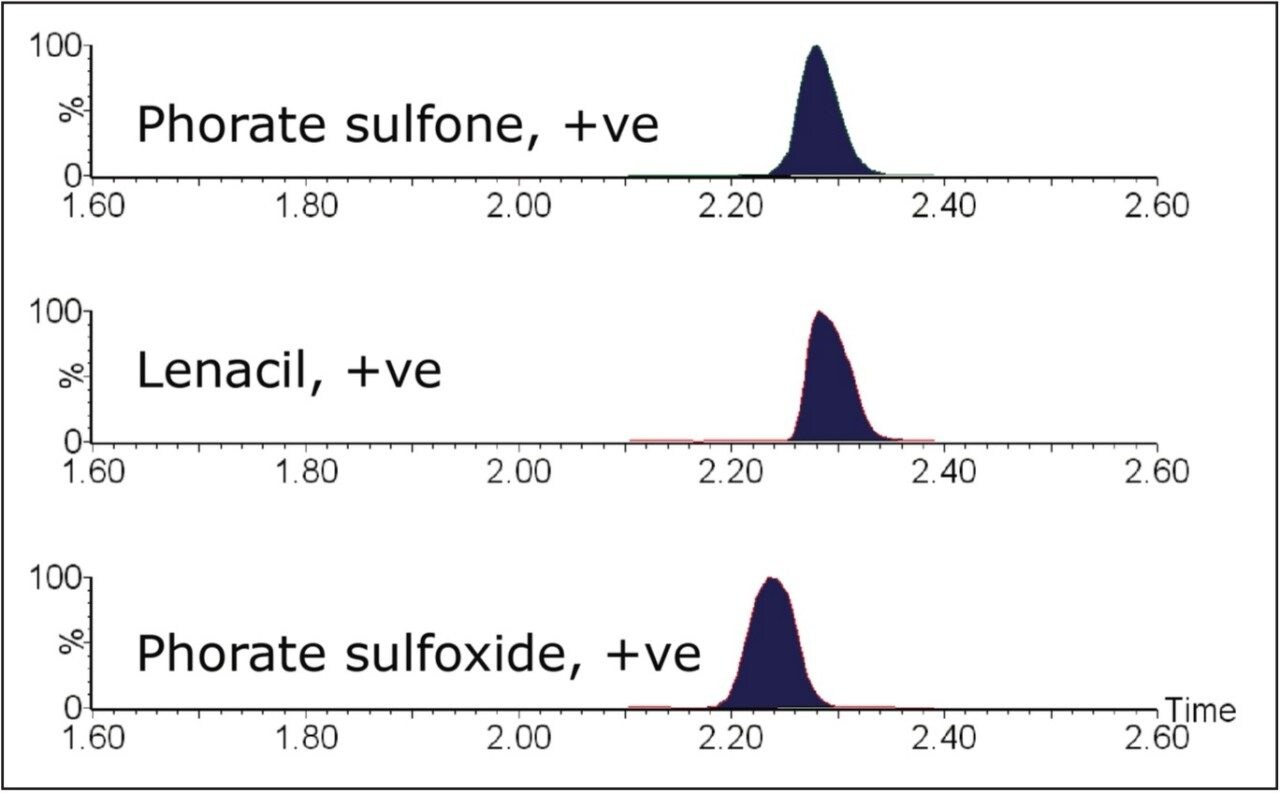
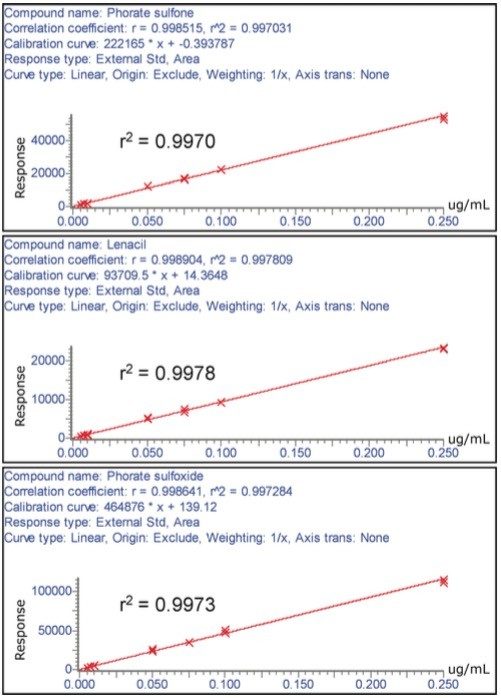
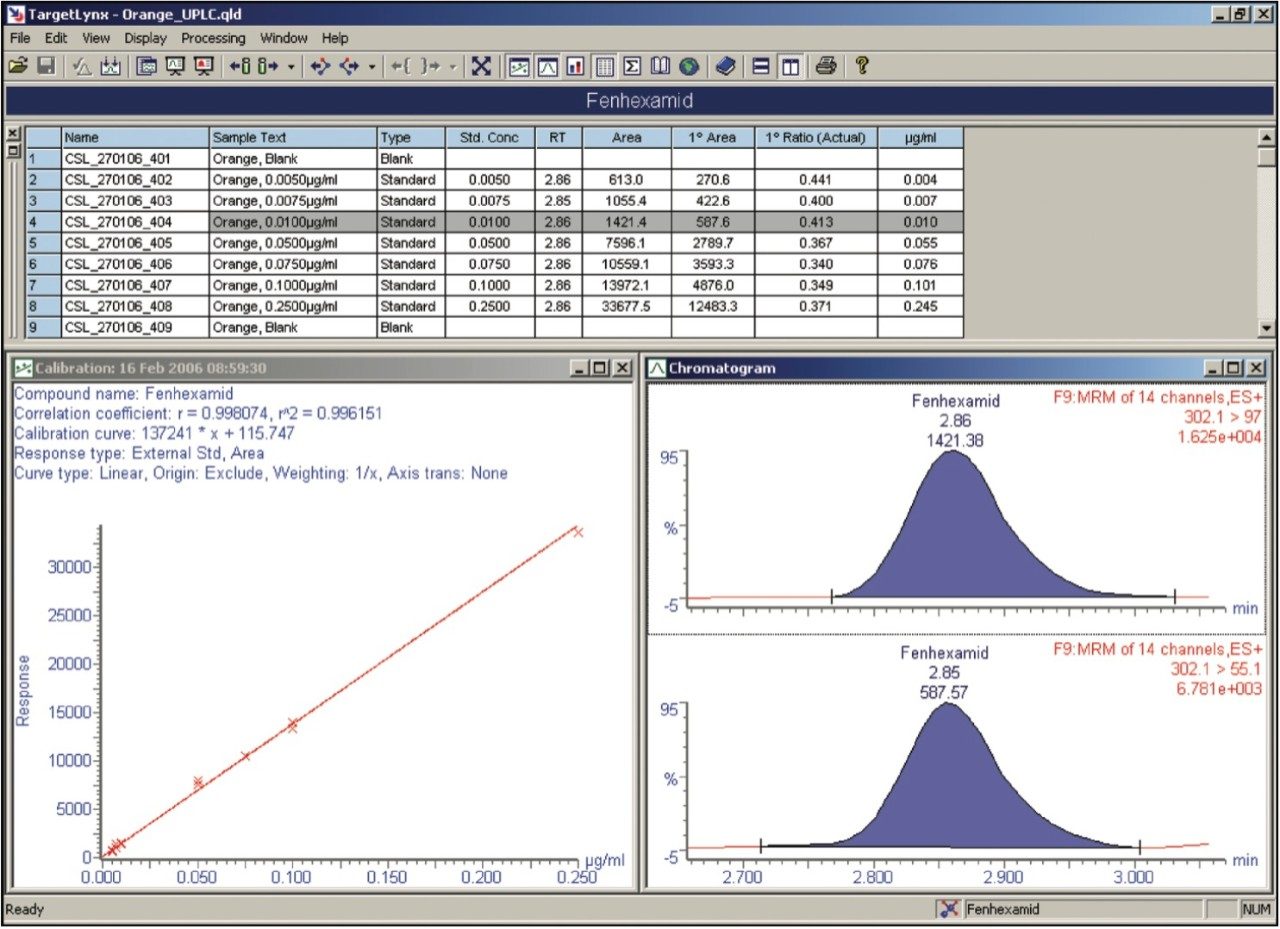
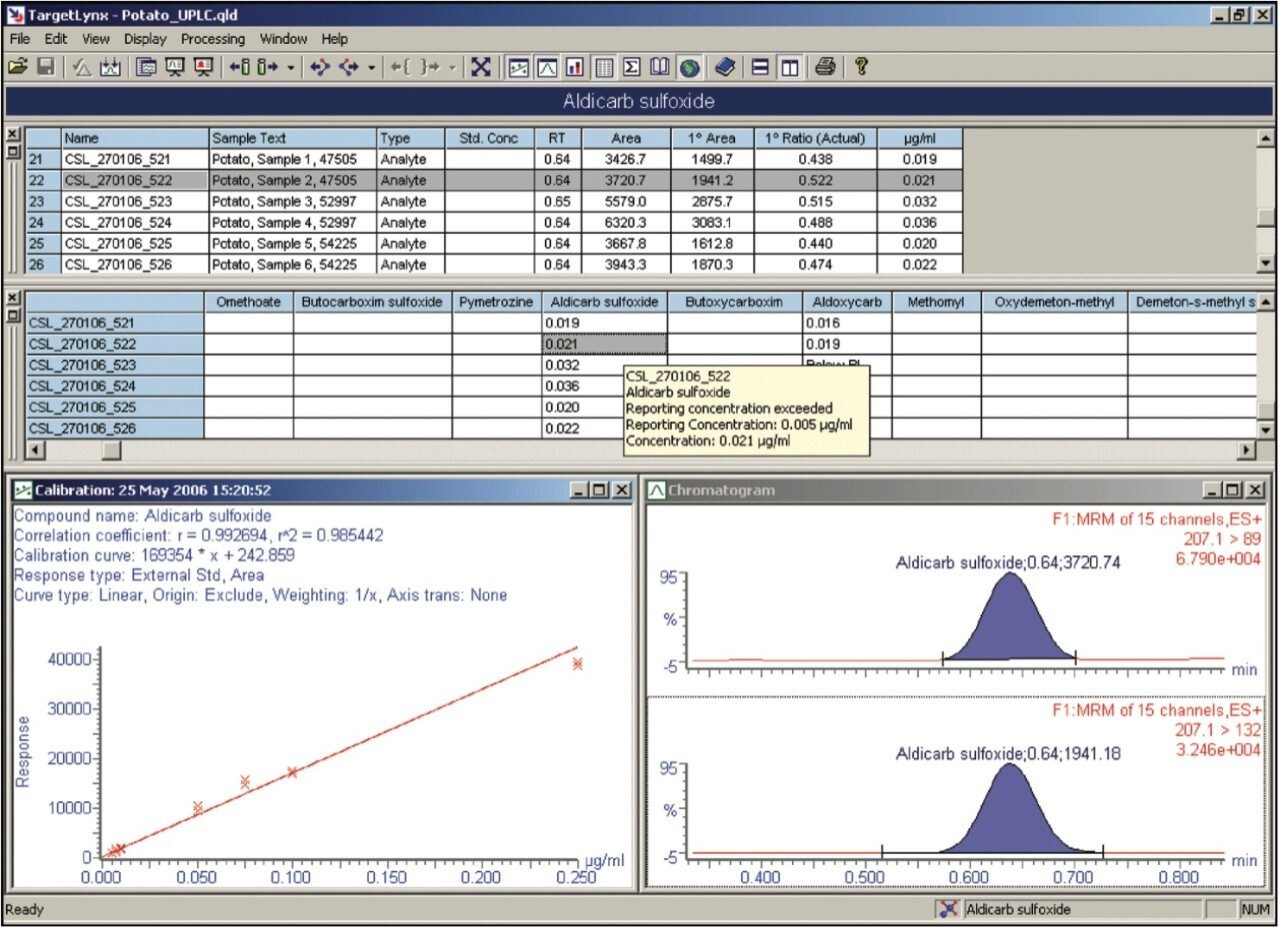
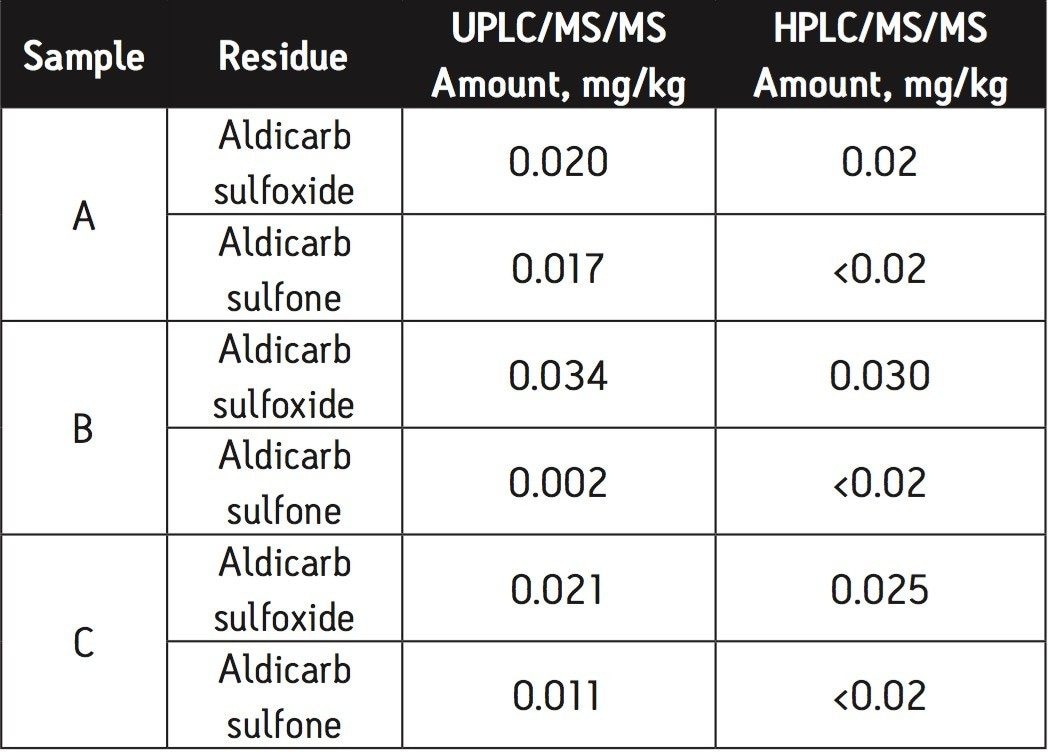
The European Union residue monitoring program 2005–2007 establishes the need to cover 55 active ingredients in various foods, including potatoes, oranges, and baby foods.1 Twenty of these pesticides are suitable for multi-residue LC-MS analysis. The majority of this group has a positive polarity in electrospray mode and only one (fludioxonil) has a negative polarity, normally requiring two injections (one in each polarity ion mode). Consequently, compounds with negative polarity are often excluded from monitoring programs. Ideally, these should be determined in a single analysis with polarity switching.
Furthermore, chemists analyzing pesticide residues are under increasing pressure to broaden the range of pesticides determined in a single analysis, to improve limits of detection, precision and quantitation, to increase confidence in the validity of residue data, to provide faster methods, and to reduce usage of hazardous solvents while maintaining or reducing costs. In order to meet these demanding requirements the scope, sensitivity, efficiency, and speed of multi-residue methods of analysis must be improved.
Given that there are many active ingredients used to control pests, it is often advantageous to extract and determine as many of them as possible during a single analysis. An extraction, with acetonitrile, followed by dispersive solid phase extraction (SPE) clean-up has been reported for the analysis of a wide range of pesticides in fruits and vegetables2 and fatty samples.3