Flavonoids are a remarkable group of plant metabolites that ubiquitously exist in natural products that have been considered as an active ingredient of many medicinal plants.1 Generally, the backbone of flavonoids consists of two phenyl rings and a heterocyclic ring, but they are often conjugated to a carbohydrate moiety with individual differences arising from various chemical processes, such as hydroxylation, methoxylation, glycosylation, and acylation.2
Plants containing flavonoids have been used for thousands of years in traditional Eastern medicine. In recent years, plant flavonoids have been shown to be of vital significance to humans. They have been linked as active contributors of health benefits, including its antioxidant properties in beverages such tea and wine, and in foods such as fruits and vegetables.
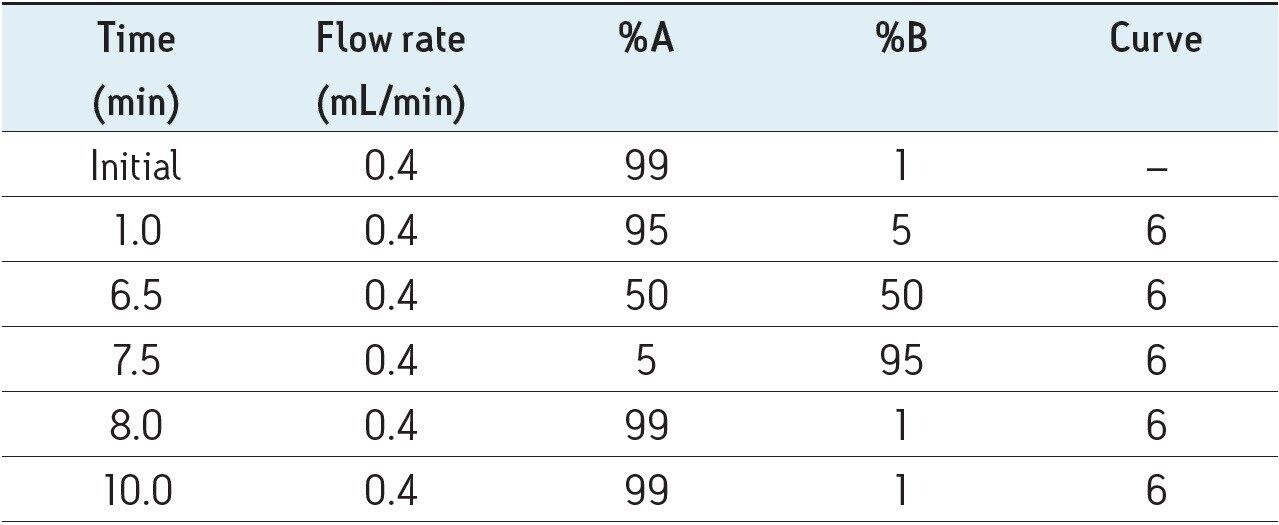
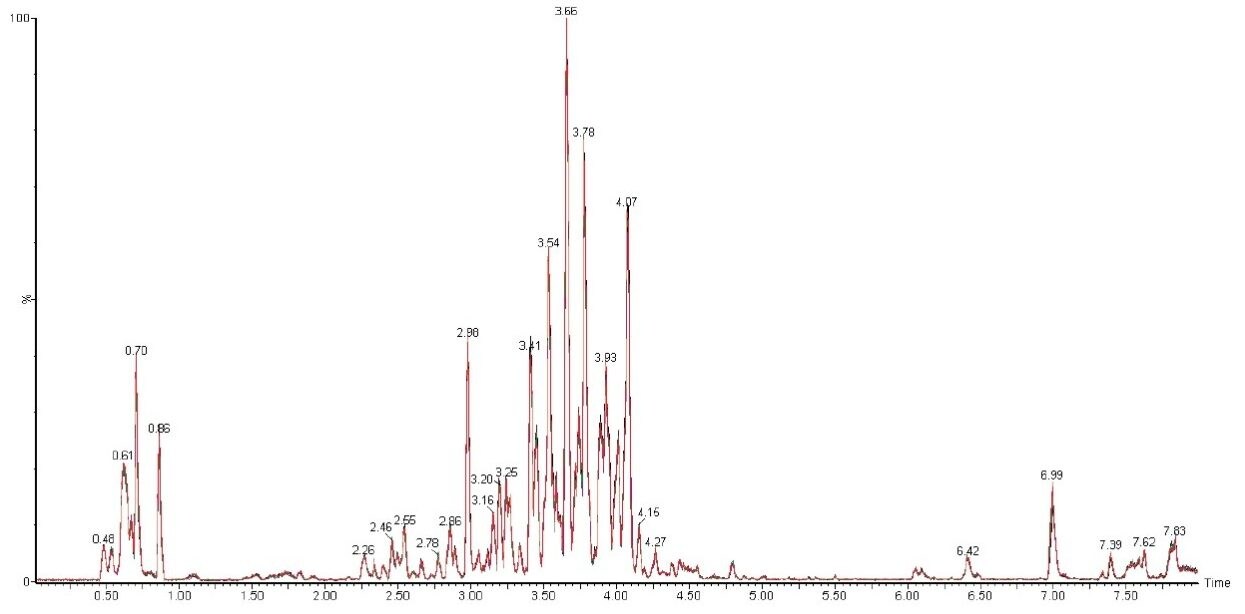
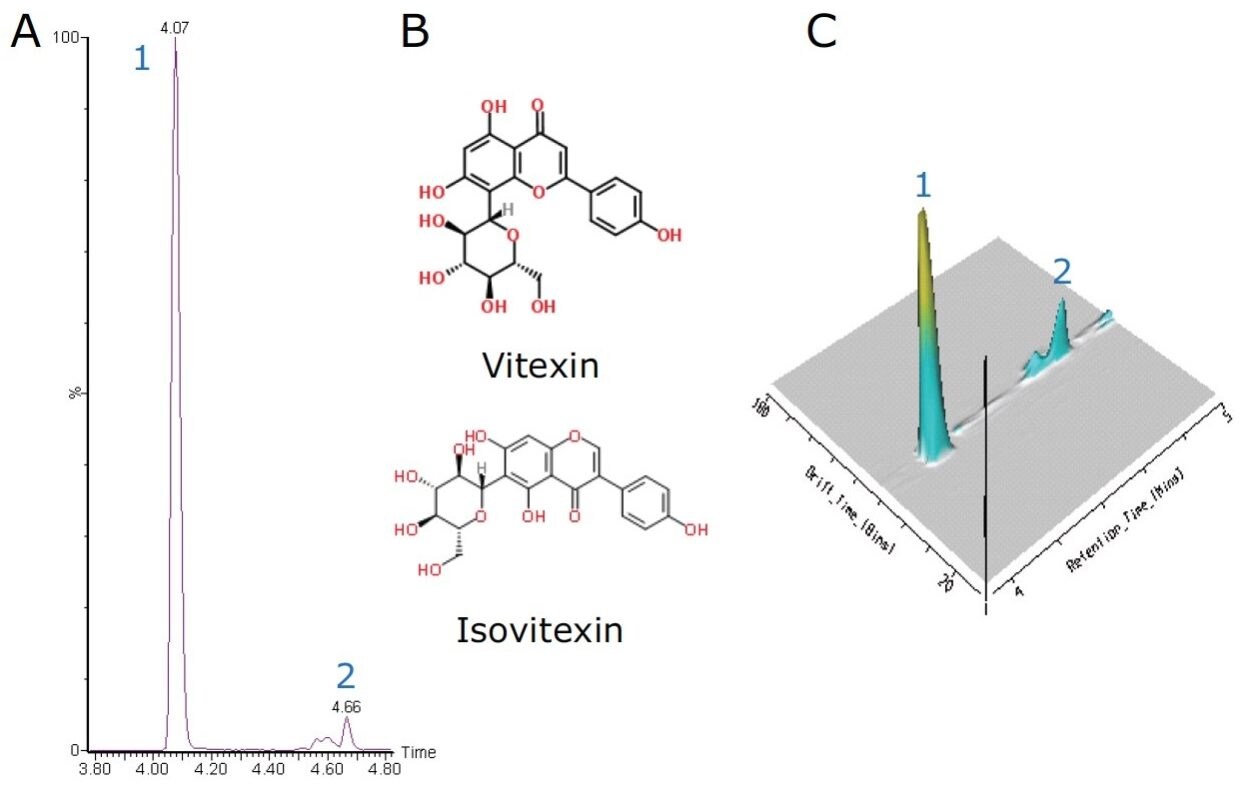
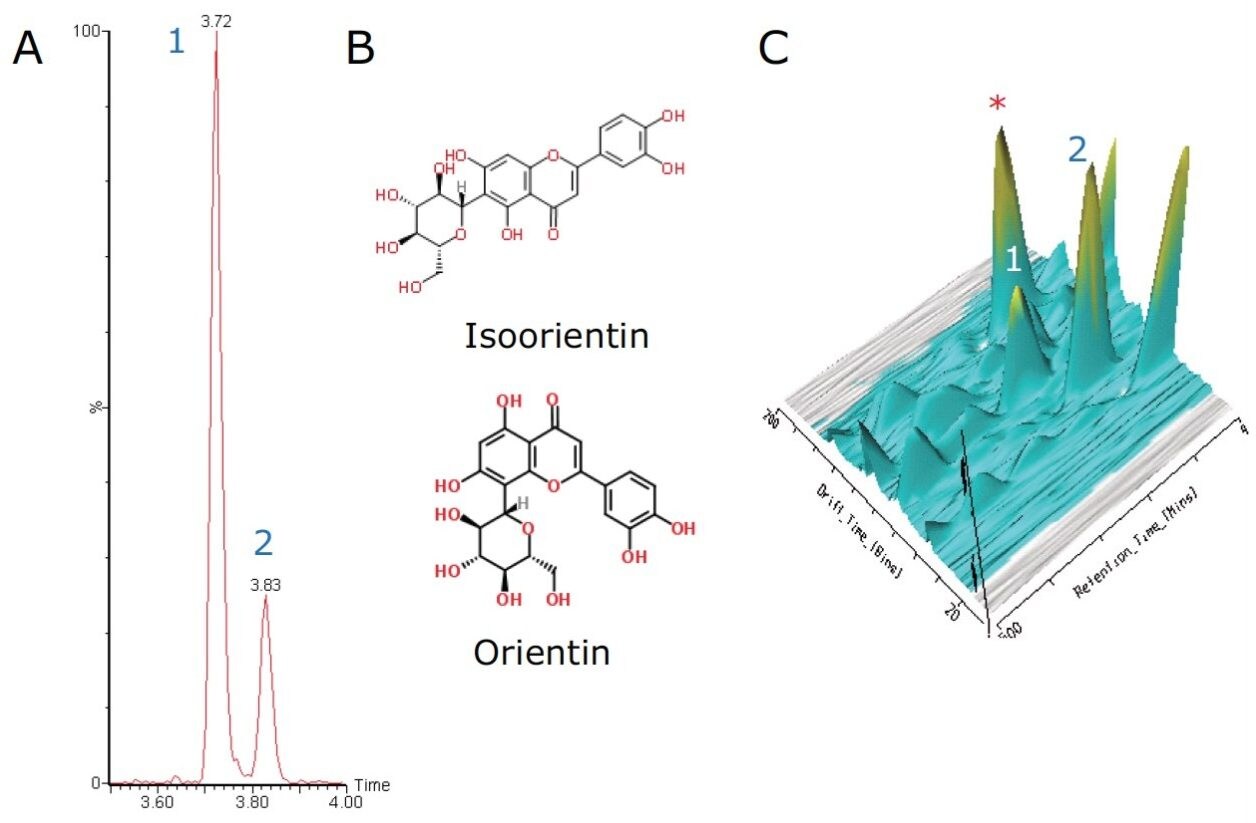
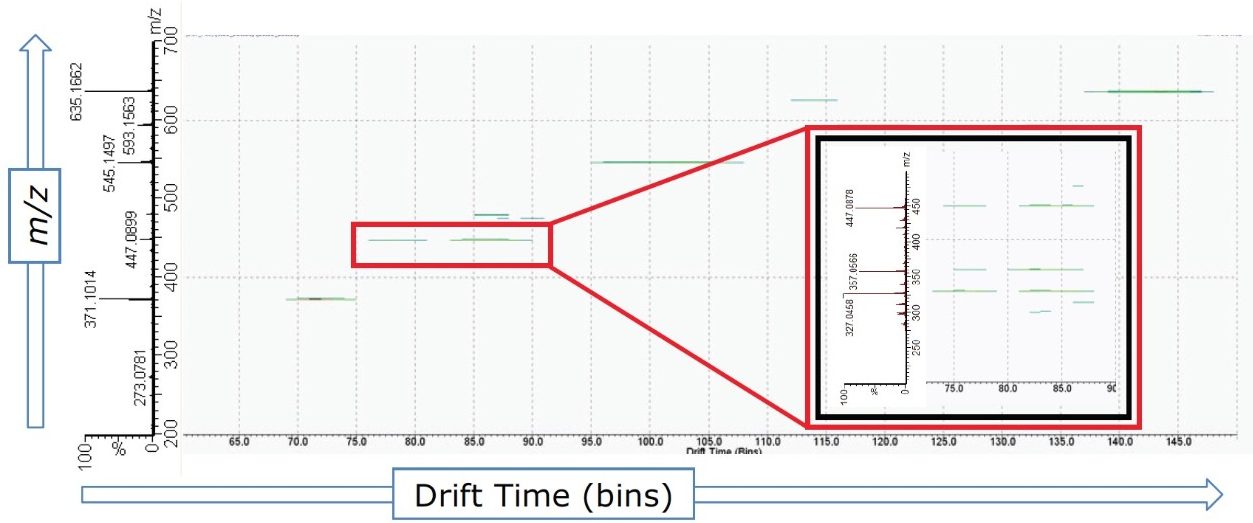
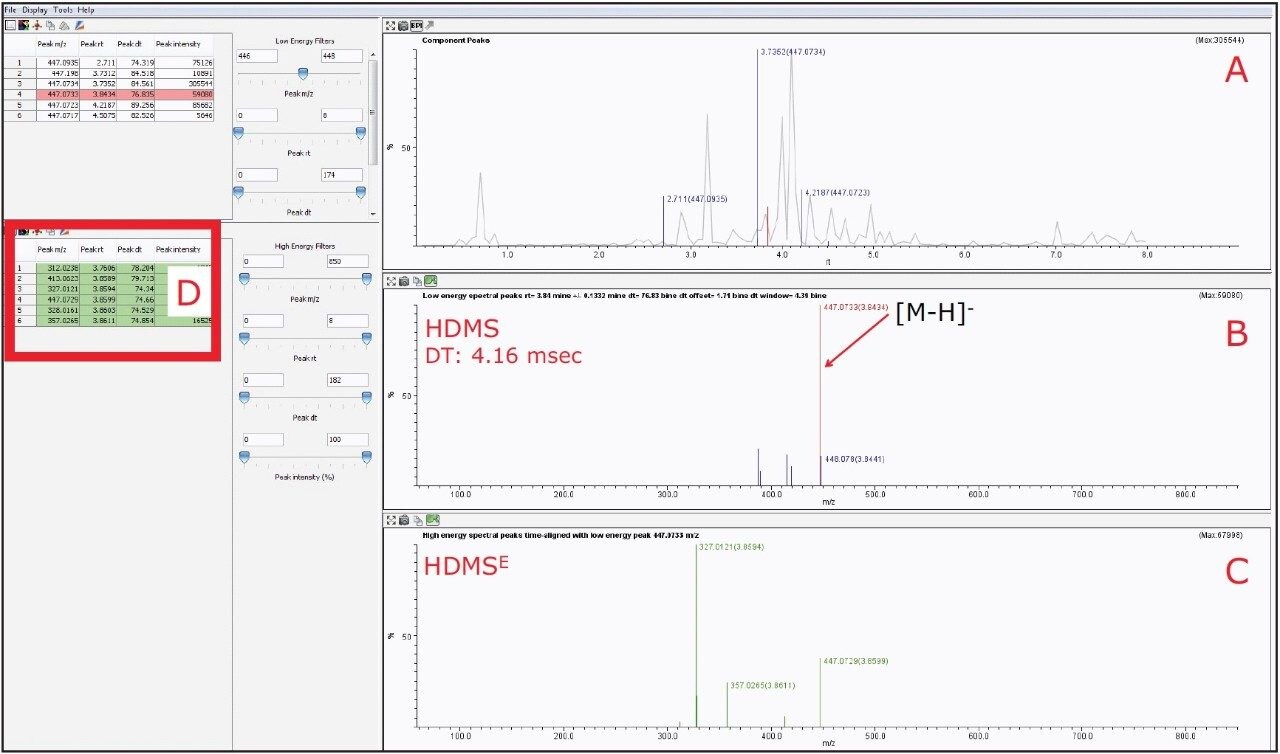
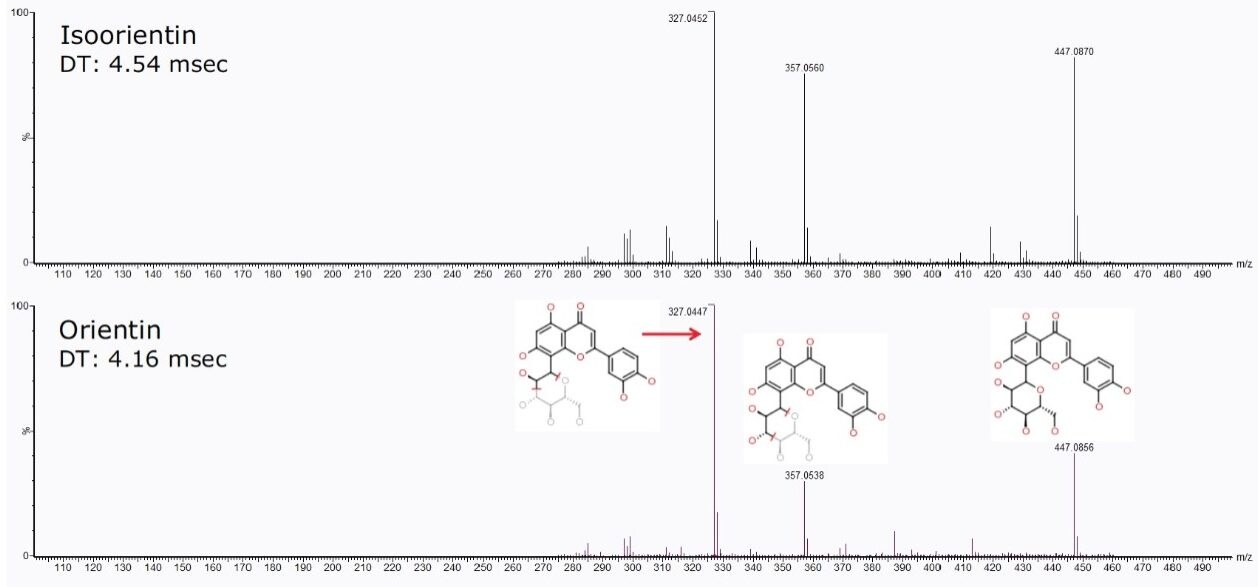
Waters SYNAPT G2 High Definition Mass Spectrometry (HDMS), a combination of high-efficiency ion mobility separation (IMS) and time-of-flight (TOF) mass spectrometry, has been used to effectively separate and identify natural product structural isomers.3 The rapid orthogonal gas separation technique in the IMS T-Wave allows another dimension of separation via their mass and shape without compromising MS data quality or sensitivity.
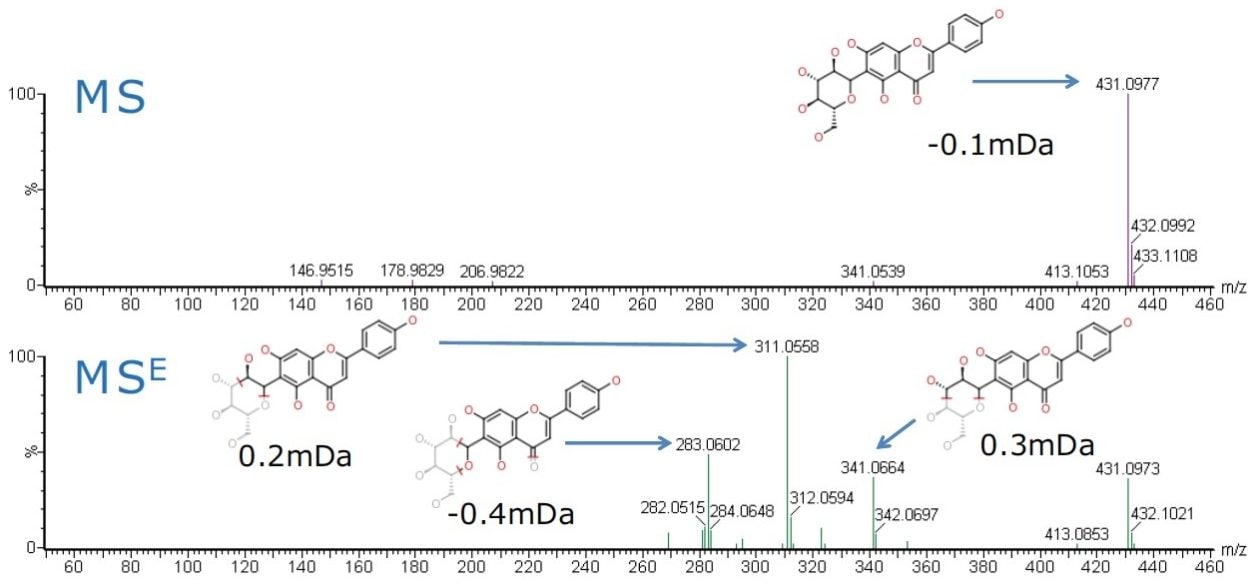
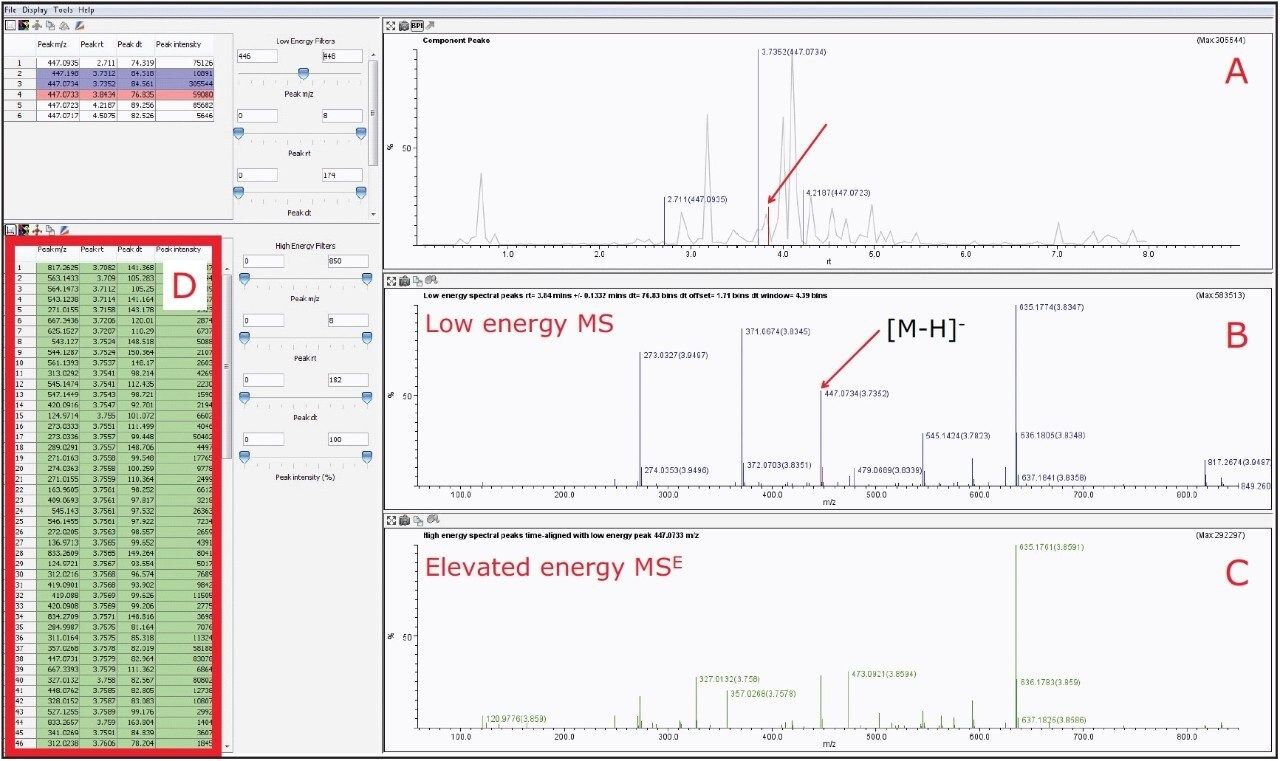
MSE is an acquisition technique that provides a simple, unbiased, and parallel route to deliver exact mass, low energy precursor (MS) and high energy fragment ion (MSE) information from every detectable component, without the need for multiple injections.
This application note describes the analysis of Ficus sp. extract using Waters ACQUITY UPLC System combined with the SYNAPT G2 HDMS System with IMS and MSE functionality to provide chromatographic and isobaric separation for a more comprehensive structural characterization of flavonoids.