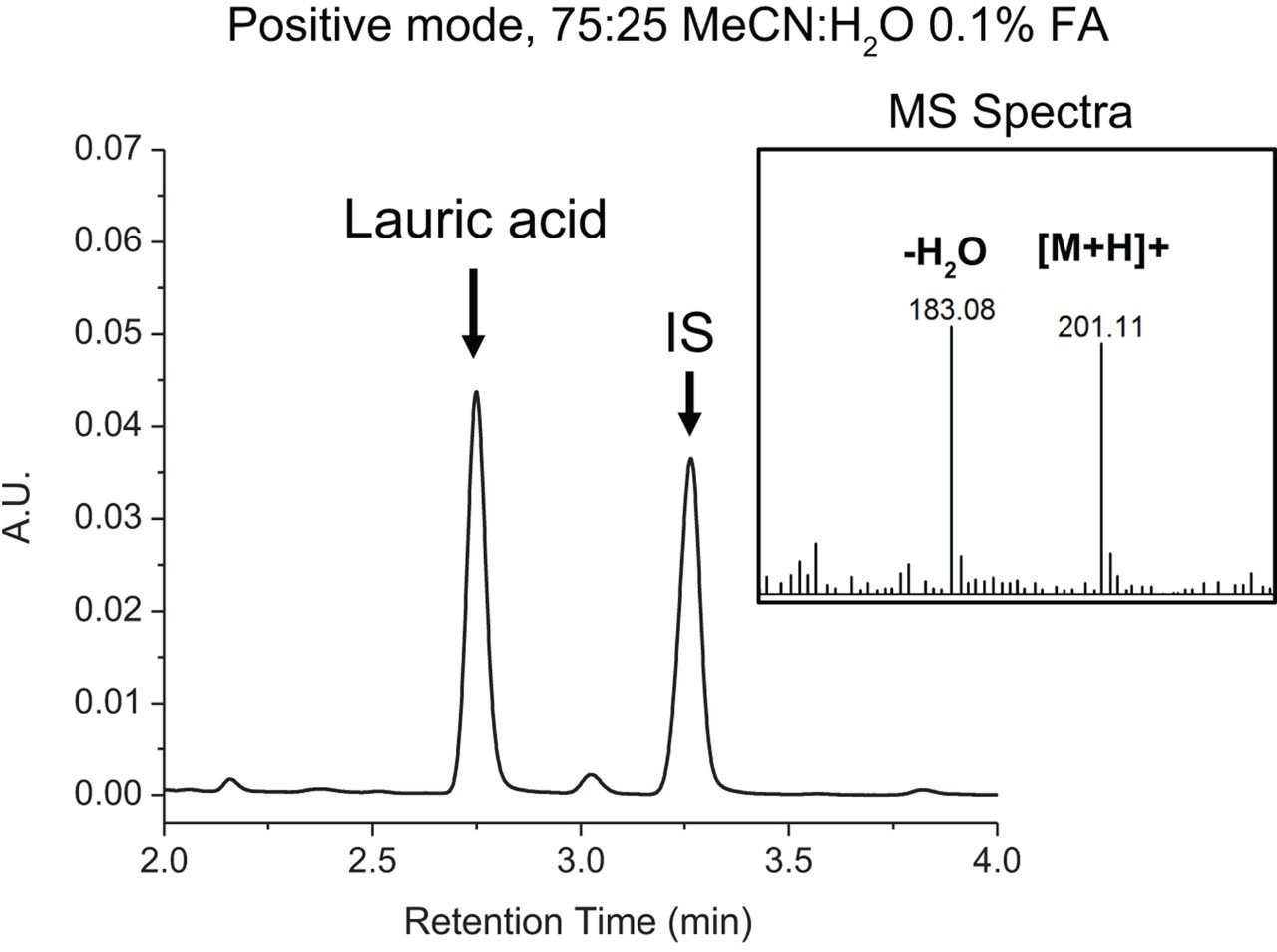
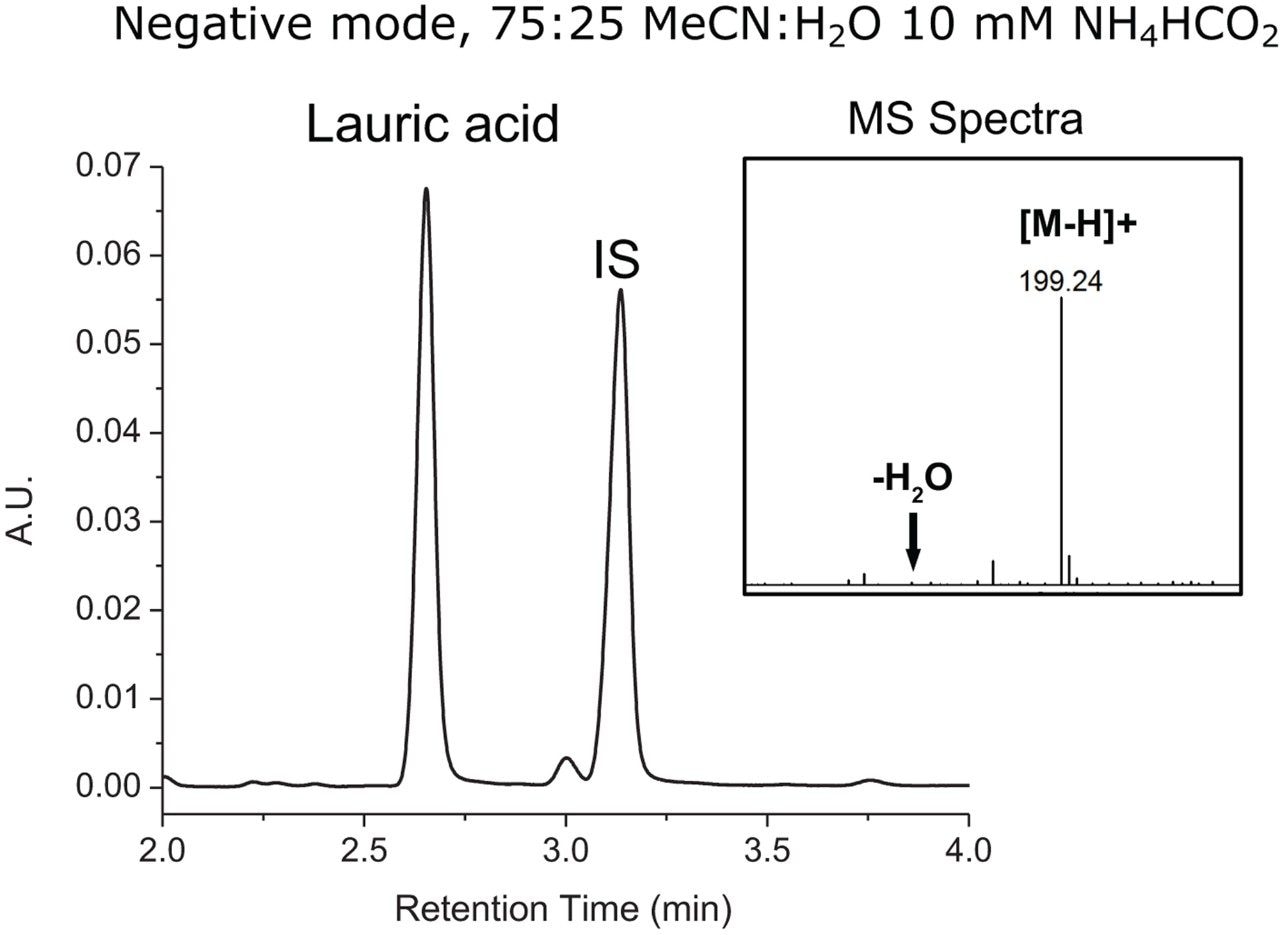
Structural considerations of analytes under investigation play a critical role in the selection of mobile phase conditions and instrument settings in LC-MS-based analyses. Reversed-phase chromatography is well suited in the analysis of FFAs when considering their hydrophobic properties, however conventional RPLC mobile phases using acidic modifiers tend to induce water loss in the MS source when acquiring data in positive mode as the carboxylic group can readily accept protons. Given their structural properties, MS acquisition in negative mode is fundamentally more appropriate for fatty acids analysis as water loss is not thermodynamically favorable. To this end, a conventional RPLC-based mobile phase comprised of 75:25 MeCN:H2O, 0.1% FA, pH ≅ 3.0 with MS data acquired in positive mode was compared against a mobile phase containing 75:25 MeCN:H2O, 10 mM NH4HCO2, pH ≅ 7.0 with MS data acquired in negative mode. Lauric acid and an internal standard (IS) were prepared at 0.1% w/w and analyzed under optimized RPLC conditions using an ACQUITY UPLC BEH C8 Column (130Å, 1.7 μm, 2.1 mm x 100 mm, p/n 186002878) on an ACQUITY UPLC H-Class Bio PLUS System. Samples were run using an isocratic method with a flow rate of 0.2 mL/min at 30 °C. The ACQUITY QDa Mass Detector was configured in-line to evaluate spectral response. As shown in Figure 1, chromatographic performance of lauric acid when using 0.1% FA was acceptable with minimal tailing and a resolution of 5.63 from the internal standard. However, as shown in the MS spectra, peak splitting due to water loss was observed at approximately 50%. As shown in Figure 2, chromatographic performance of lauric acid was preserved with comparable peak shape, retention time, and a resolution of 5.39 from the internal standard when using 10 mM ammonium formate in lieu of formic acid. More importantly, spectral peak splitting was not observed at an appreciable level when acquiring MS data in negative mode as shown in the figure inset. This work demonstrates that spectral response of FFAs can be improved when considering structural properties during method development to reduce in-source water loss peaks for efficient spectral interpretation.