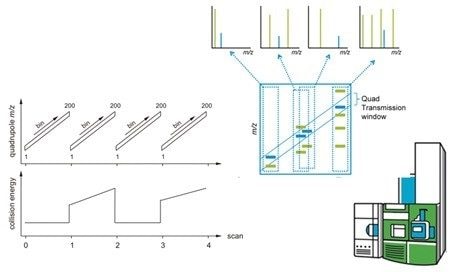
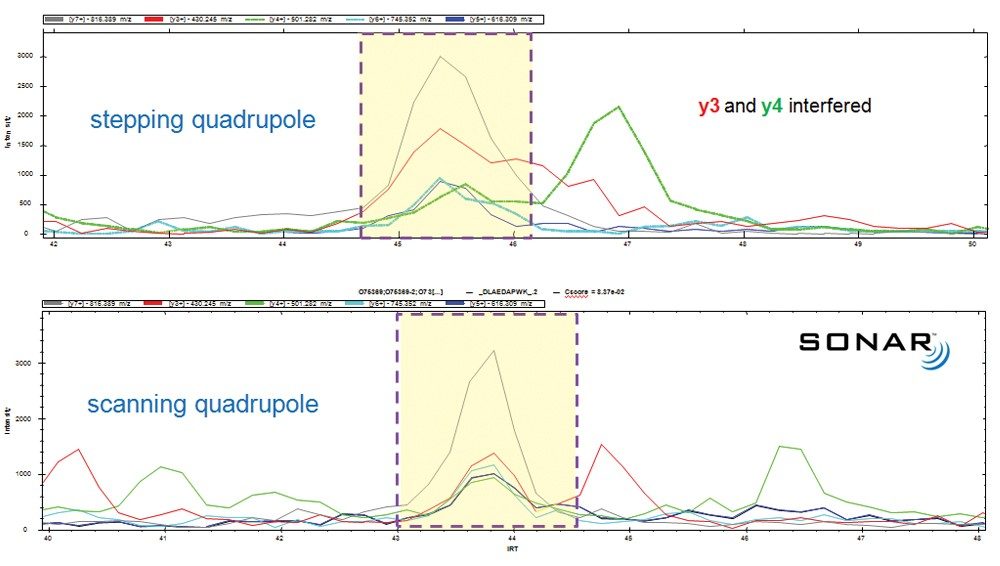
The principle of SONAR, a scanning quadrupole based data independent acquisition (DIA) method, is illustrated in the left hand side image of Figure 1. In short, alternate datasets are acquired in low (MS1) and elevated (MS2) collision energy mode.1,2 During each low and elevated energy segment, the quadrupole isolation window is scanned linearly between two user-selected positions and 200 TOF spectra are acquired. The quadrupole scan duration is application/chromatographic peak width dependent and typically varies from 0.1 s to 1 s. In the elevated energy mode, the collision energy can be ramped between two values, which are selected to optimize fragmentation efficiency at each quadrupole position. The selectivity of the acquisition method is illustrated by the middle image where, dependent on the position of the quadrupole, i.e. transmission m/z window, precursor (even those close in mass) and product ions can be exclusively isolated.
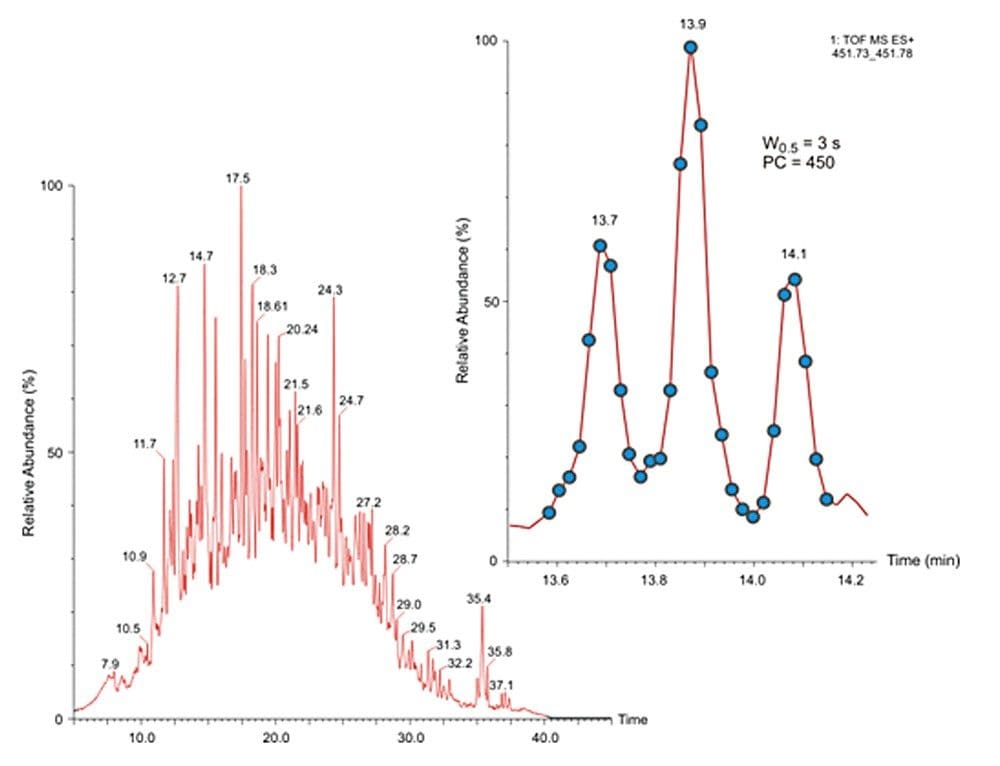
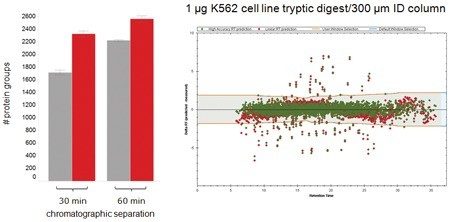
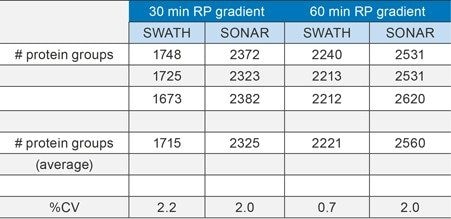
The requirement for acquisition speed is clearly demonstrated in Figure 2, showing a 50 Da wide mass extracted chromatogram for a 30 min reversed phase gradient separation of non-fractionated K562 tryptic digest. Shown inset is a 10 mDa wide mass extracted chromatogram over a narrow chromatographic window of 30 s. Typical peak widths at half height were 3 s; hence, to retain a sufficient number of points across the peaks for precise quantitation while maintaining optimum S/N, the scan speed was set to 0.5 s, providing between six and eight data points across a peak. The peak capacity for 30 min high throughput proteomic separations was estimated to be ~ 450. The importance of peak sampling frequency and its effect on quantitative precision is described in more detail elsewhere.1