Parameters of Spectrum Deconvolution
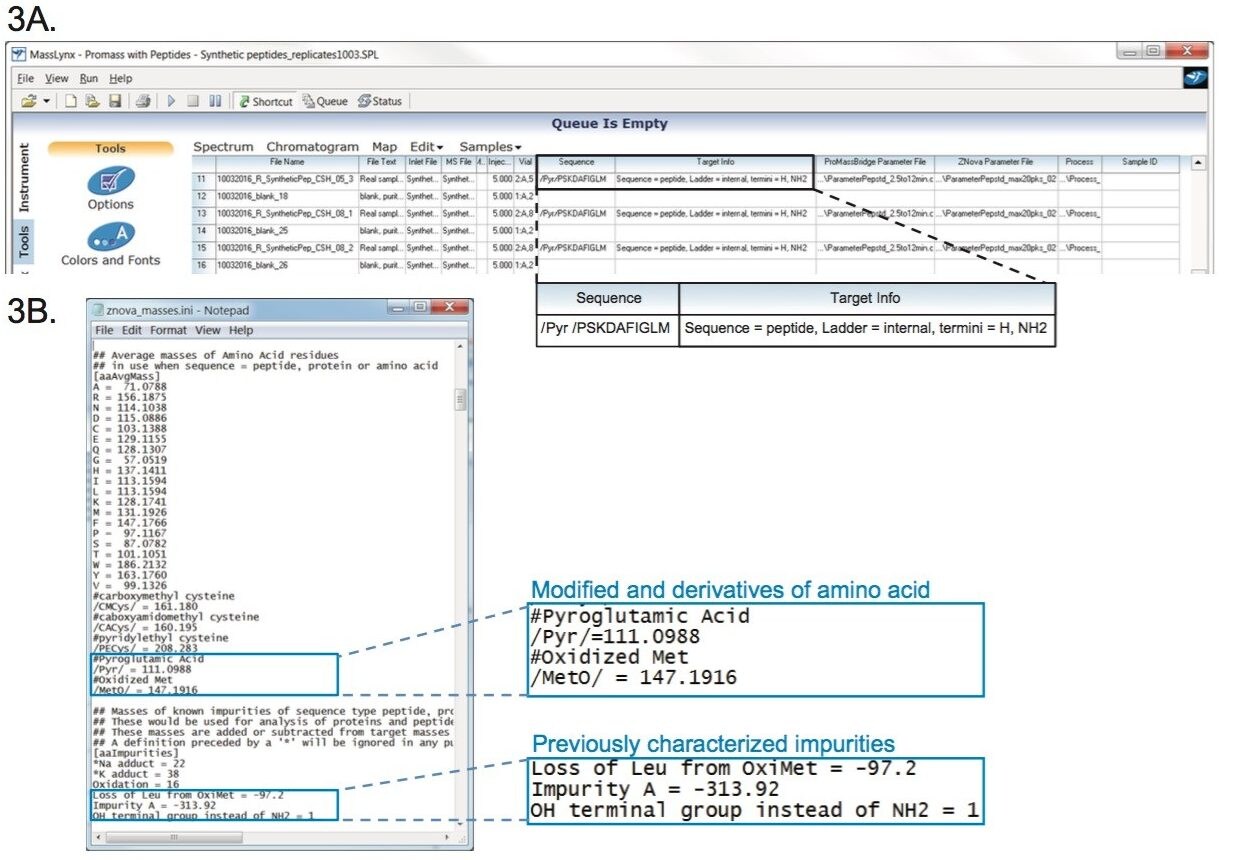
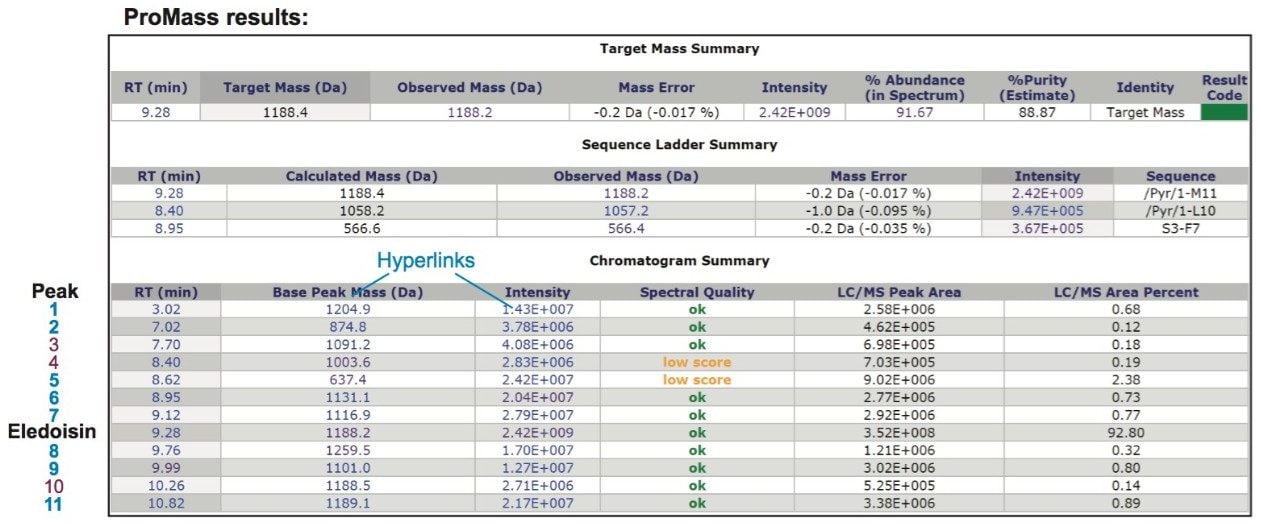
With the optimized method determined, the benefit of mass detection can now be fully utilized. The ACQUITY QDa Detector enables the software to determine mass difference between impurities and the target peptide for impurity identification. Automated data acquisition and processing of synthetic peptides can be performed by MassLynx when used in conjunction with ProMass. Figure 3A is an example of the modified Sample List in MassLynx, which contains the instrument methods for data acquisition, as well as information needed for target mass calculation, and parameter files for data deconvolution of synthetic peptides using ProMass. For peptide impurity profiling, the information of the target peptide needs to be entered in the Sequence column as well as the desired Target Info as shown in the zoom-in image of Figure 3A. In this example, ‘Ladder = internal’ was used to detect all possible fragments with either termini. The target synthetic peptide, eledoisin, has a primary amine group (NH2) at the C-terminus rather than an OH, thus requiring the termini to be specified as such for correct mass determination. Parameters for MS spectra analysis are defined by the files entered into the ProMassBridge Parameter File column and the Znova Parameter file column. The ProMassBridge Parameter File enables communication between ProMass and MassLynx, and contains user definable parameters such as the retention time window to process, smooth functions and baseline subtraction. As part of the ProMass software, the Znova™ Parameter file provides parameters for spectrum deconvolution, and thresholding for streamlining reporting.
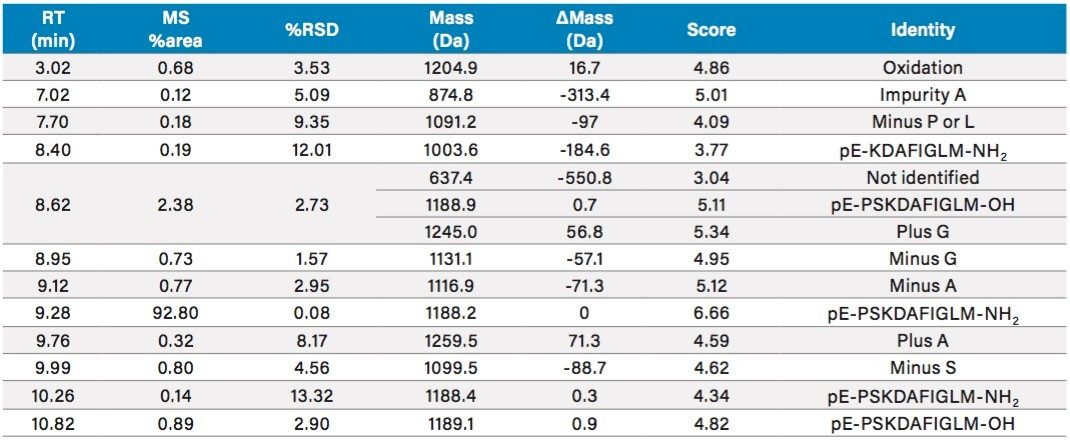
The Znova processing file contains a customizable database for target identification and impurity profiling as shown in Figure 3B. Modified and derivatized amino acids can be entered along with naturally occurring amino acids as part of the “building-blocks” for calculating target masses. In the case of eledoisin, Pyroglutamic Acid (/Pyr/=111.1) was entered to account for the amino acid derivative at the terminus of the synthetic peptide. Similarly, previously characterized impurities can be manually entered as their calculated mass differences from the target peak. As shown in the blue rectangle of Figure 3B, deletion of leucine from the oxidized target peptide (loss of Leu from OxiMet = -97.2), possible peptide fragment (Impurity A = -313.9), and C-terminal substitution (OH terminal group instead of NH2 = 1) were previously identified by high resolution MS prior to this study. Matching mass differences will be shown in deconvolution reports along with their identities.