Food allergies arise from an abnormal immunological response to certain foods. Proteins are the main candidates for triggering allergic reactions. Egg-based proteins are one of the most frequent causes of adverse reactions in food. Since many processed foods contain egg as a raw ingredient, the ability to assess changes in protein structure and detection through the manufacturing cycle is important.
Food allergen analysis using LC-MS/MS is a current hot topic for many food scientists and there are two approaches that can be used to generate a quantitative method. The first approach is to perform in silico digestion of proteins, based on fasta sequences available in databases (e.g. UniProt), providing a list of potential peptides and MRM transitions. This methodology requires further investigation to determine which MRMs are the most specific and sensitive at detectable response levels for post-food processing, sample treatment, and during the ionization process. The alternative is to perform a discovery omics experiment using a high resolution instrument, such as a QTof mass spectrometer and use the data observed from this experiment to generate a targeted method.
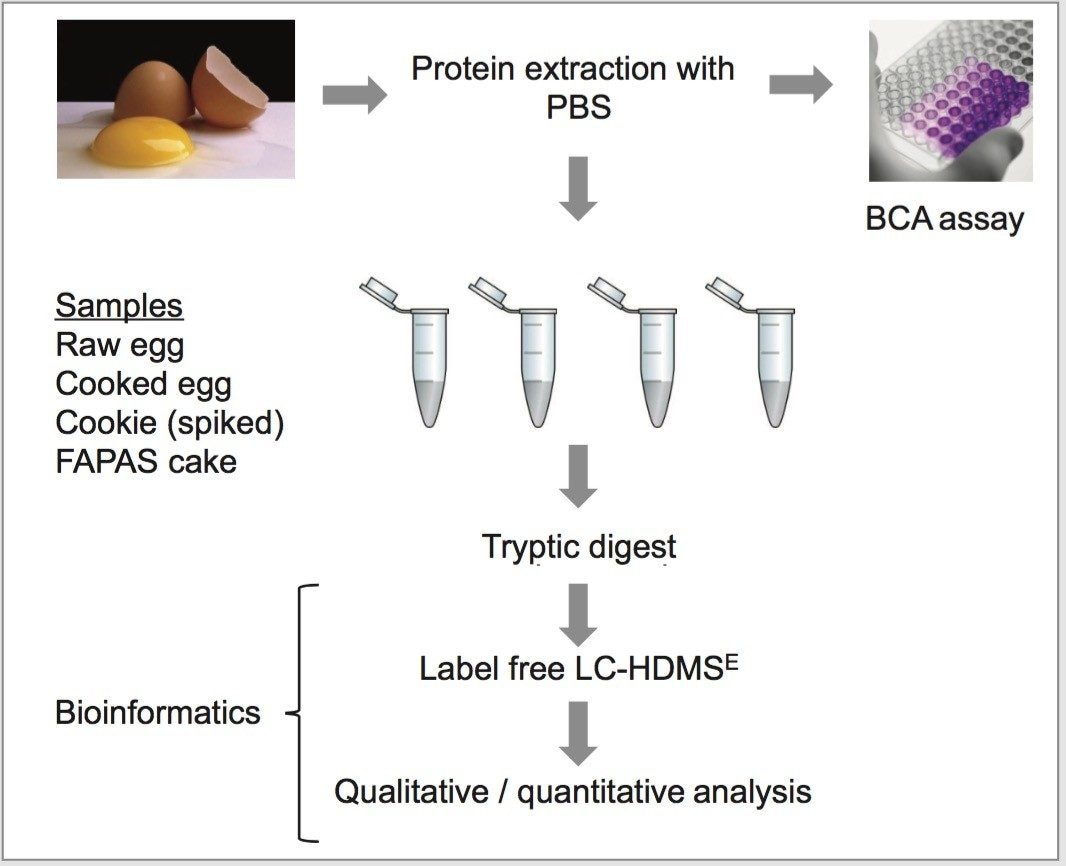
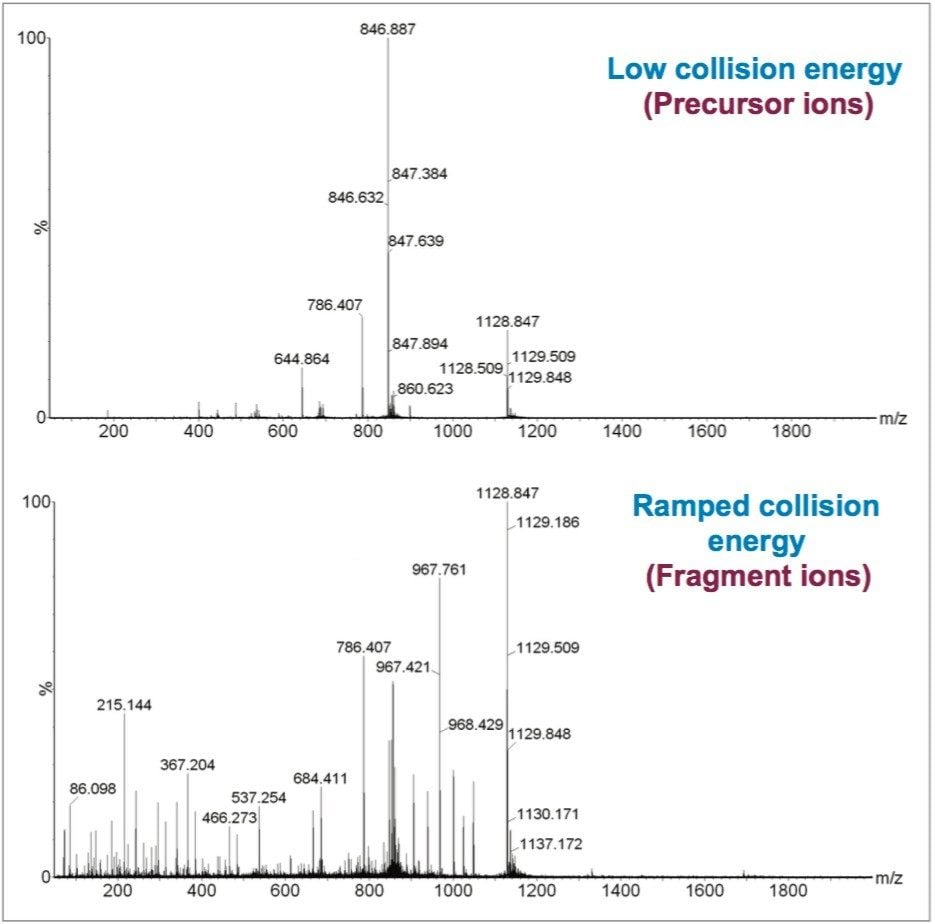
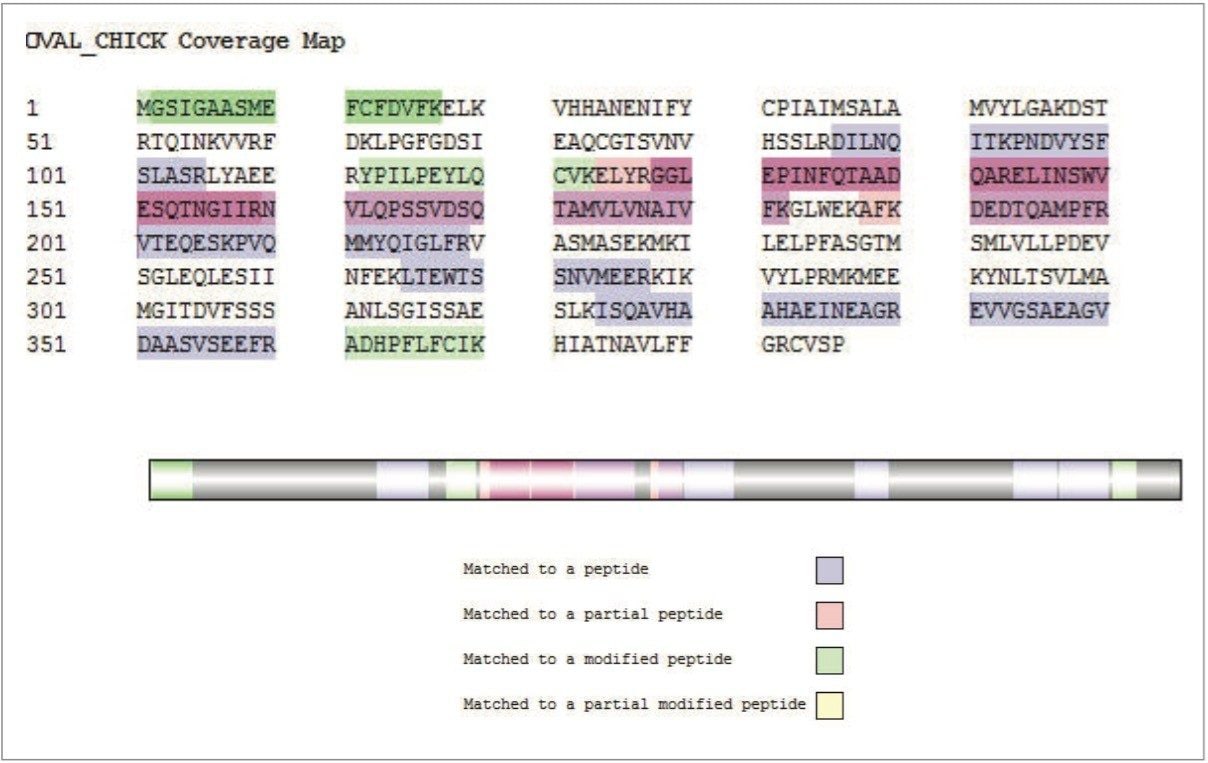
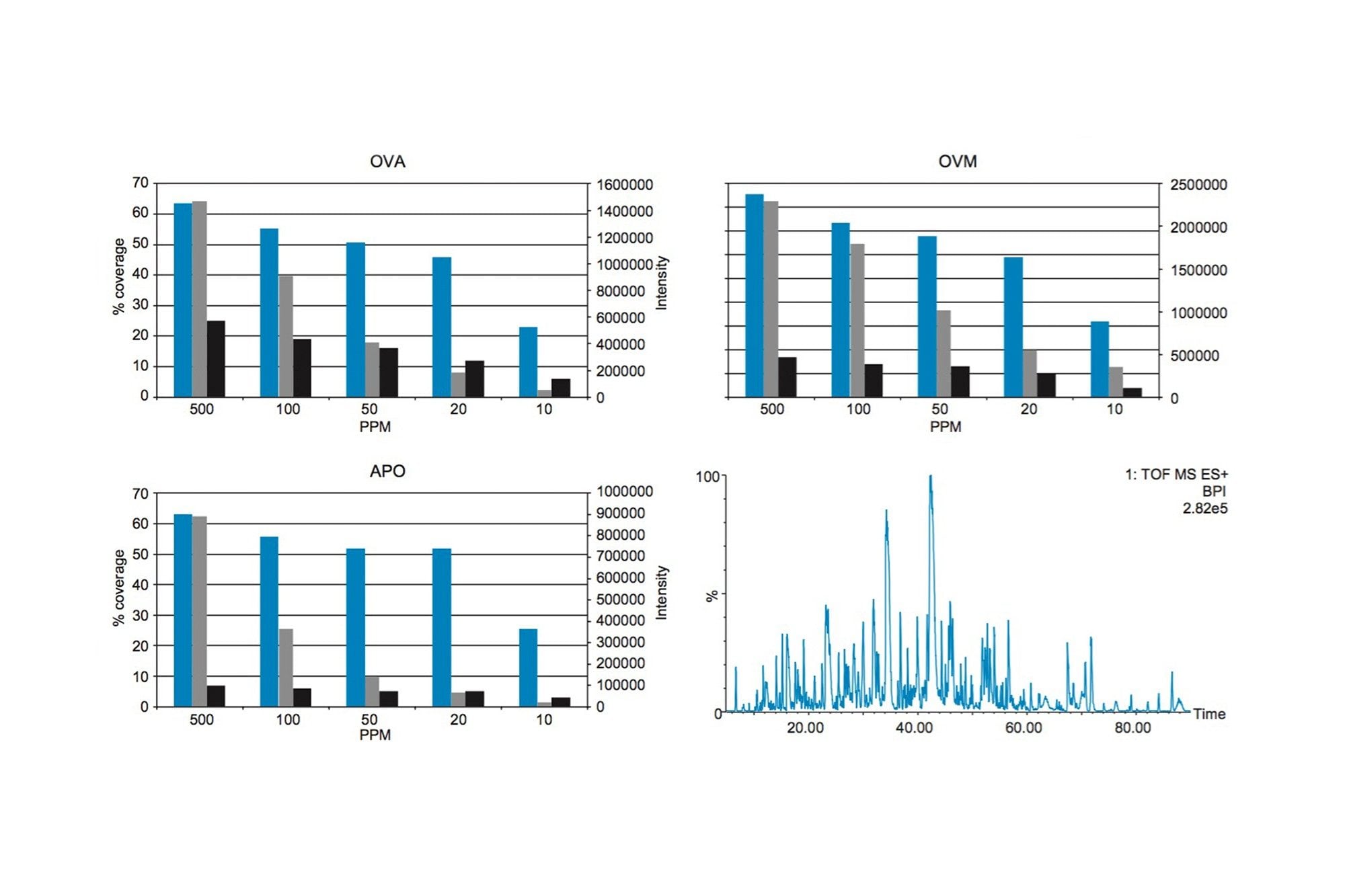
In this study, the second approach has been applied and focuses on identifying and quantifying known allergenic proteins from raw and cooked egg samples. Proteins extracted from raw and cooked egg samples were digested using trypsin and label-free protein expression data were acquired with Waters SYNAPT G2-Si using an ion mobility data independent approach (whereby the collision energy was switched between low and elevated energy states during alternate scans).
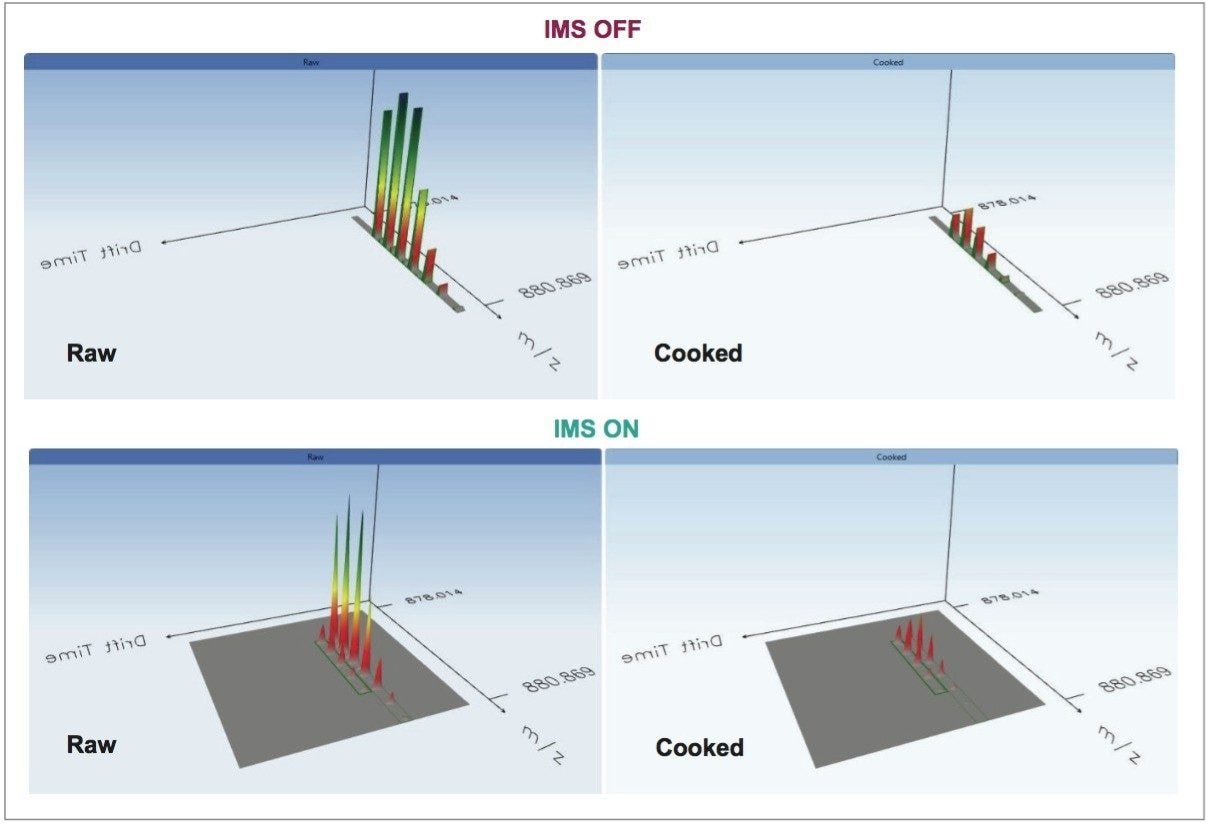
Utilizing ion mobility as part of the workflow provides enhanced specificity and therefore confidence of identifications returned, even in the presence of complex matrices, such as processed food samples. Precursor and product ions were associated by means of retention and drift time alignment. Although egg proteins were the focus of this work, other allergenic proteins that are also extracted using the sample preparation could be investigated, providing a potential means for multi-allergen detection.
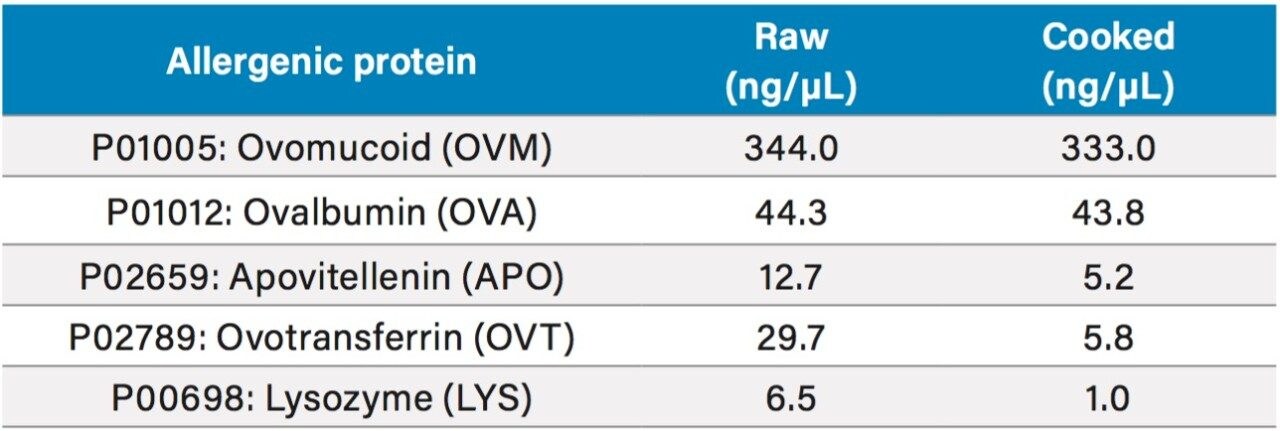
The acquired data were processed using Progenesis QI for Proteomics and searched against a Gallus Gallus (Uniprot) database. The results generated allowed for relative quantification to be established. The results of this study showed that a significant proportion of proteins identified were expressed when comparing cooked and raw egg sample sets, which included known allergenic proteins (e.g. apovitellenin I). Peptides identified in both sample sets allowed for MRM transitions to be generated and a quantifiable value assigned.