Amongst forensic toxicology laboratories, cannabis is one of the most frequently encountered drugs.1 For most forensic laboratories providing toxicological analyses, the confirmation, and quantification of Δ9-tetrahydrocannabinol (THC) and its primary metabolites in whole blood or plasma is high on the list of requests from clients and enforcement agencies, such as district attorneys and/or police departments (city, county, or state levels).2 While whole blood is often used in postmortem forensic cases, whole blood/plasma ratios can be quite inconsistent, particularly for COOH-THC.3 Therefore, for pharmacokinetic studies involving medicinal uses or behavioral effects, plasma is often a preferred matrix.4,5 The ability to efficiently extract and quickly analyze samples submitted by such agencies is seen as a definite benefit to the laboratory. In this application, the efficient extraction and accurate quantification of THC and two major metabolites, THC-COOH and THC-OH, is achieved using a novel solid-phase extraction (SPE) sorbent, Oasis PRiME HLB, coupled with fast analysis using LC-MS/MS. This method offers a great solution for laboratories looking to incorporate this type of analysis into their methodology portfolio.
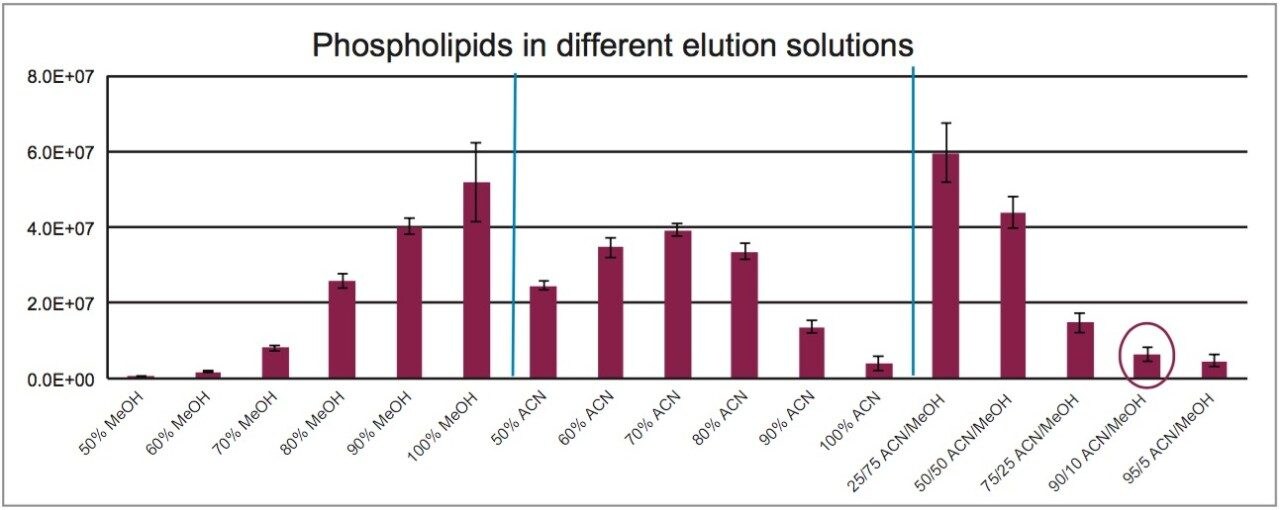
Sample preparation is an important consideration for forensic toxicology laboratories. Specifically, there is a desire to use the simplest possible method which yields the maximum cleanup. THC and its metabolites, in particular, can be prone to non-specific binding during sample preparation and manipulation, so any method that minimizes sample manipulation, evaporation, and reconstitution is desirable. This application employs Waters’ newly developed sample preparation sorbent, Oasis PRiME HLB, which is designed to provide several key advantages over traditional SPE sorbents. These include the ability to eliminate sorbent preconditioning and equilibration, creating a faster workflow compared to traditional SPE products. It also has the ability to remove more matrix interferences, particularly phospholipids, resulting in cleaner extracts and reducing the risk of short column lifetimes or MS source fouling. Compared to liquid-liquid extraction (LLE) methodologies, this method avoids the use of toxic, LC incompatible solvents and does not require evaporation and reconstitution of the extracts. In addition, phospholipids are generally not removed by LLE, so the extracts produced from this method are cleaner in that respect, with less risk of ion suppression from residual phospholipids.
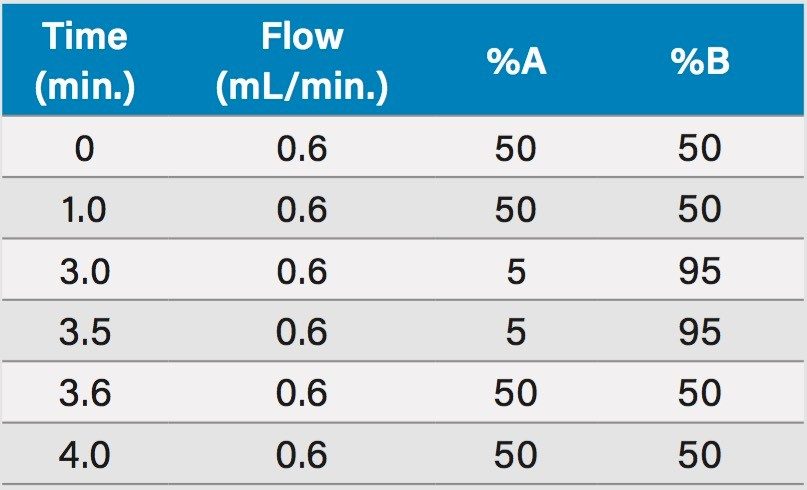
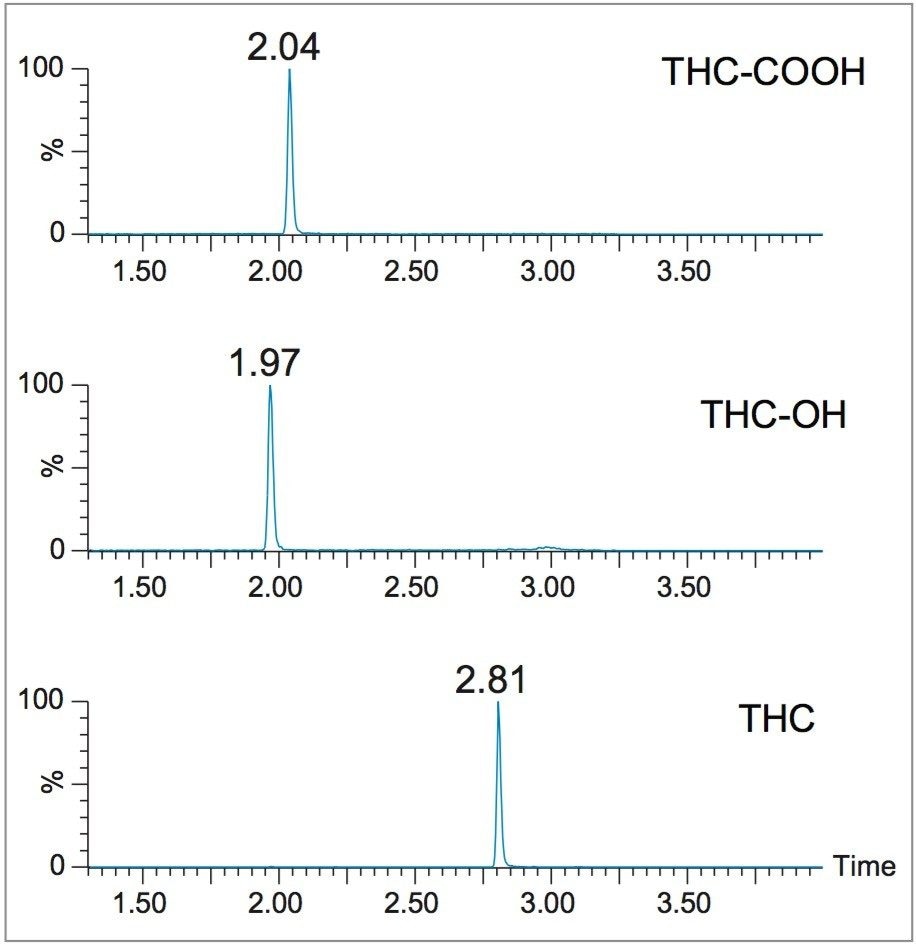
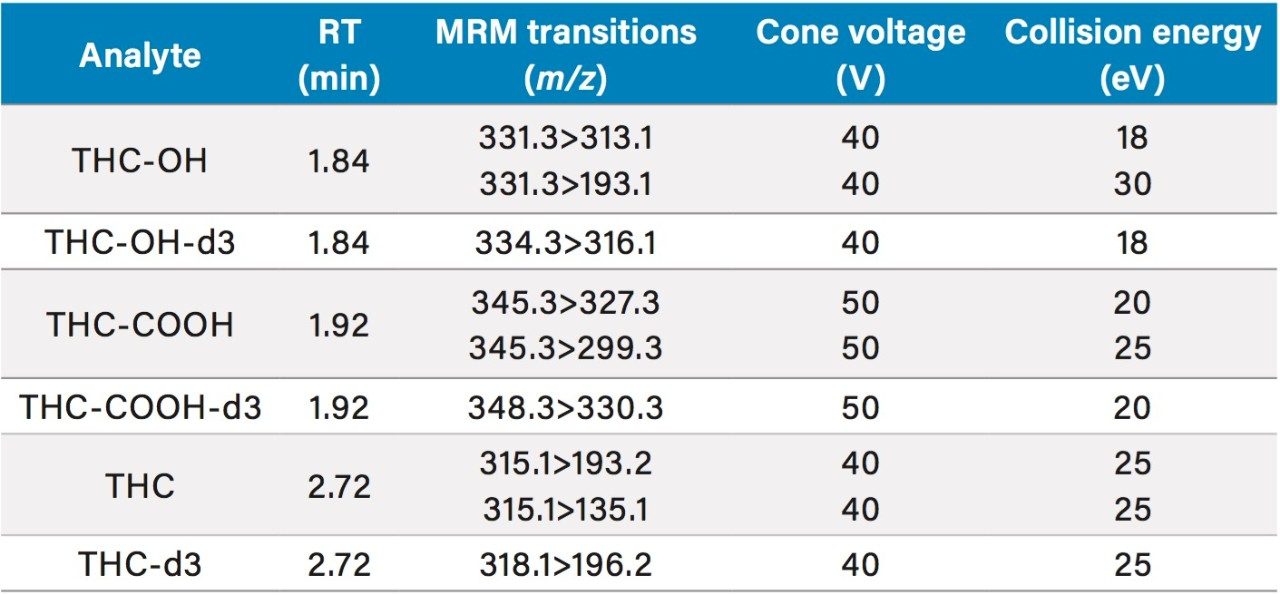
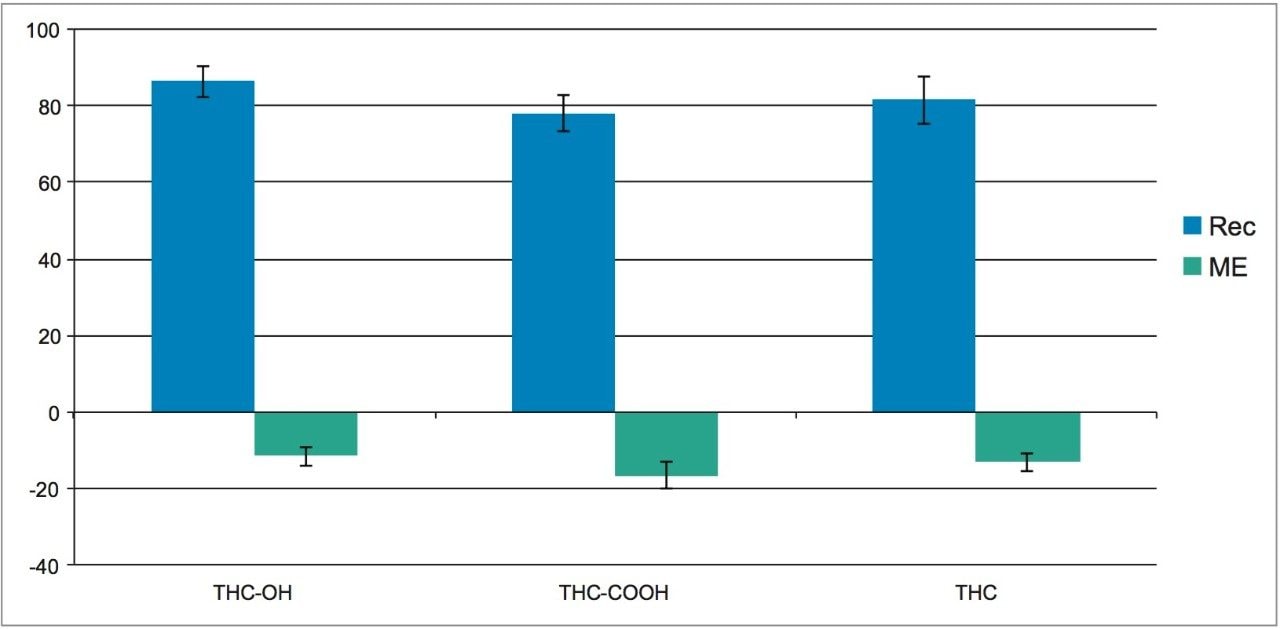
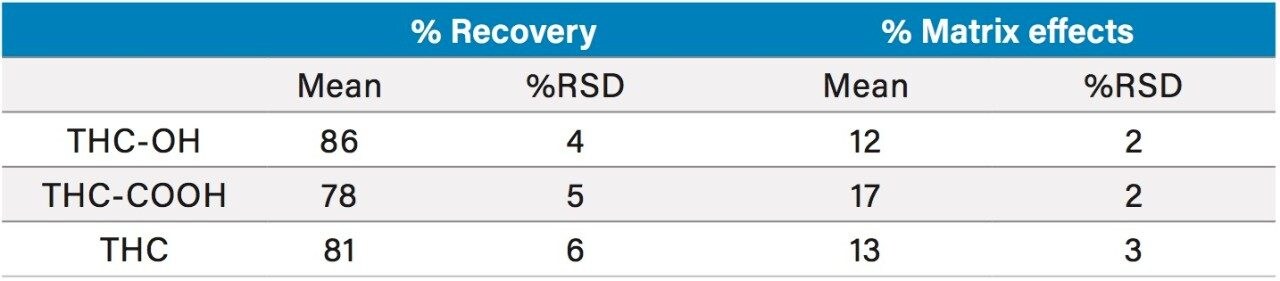
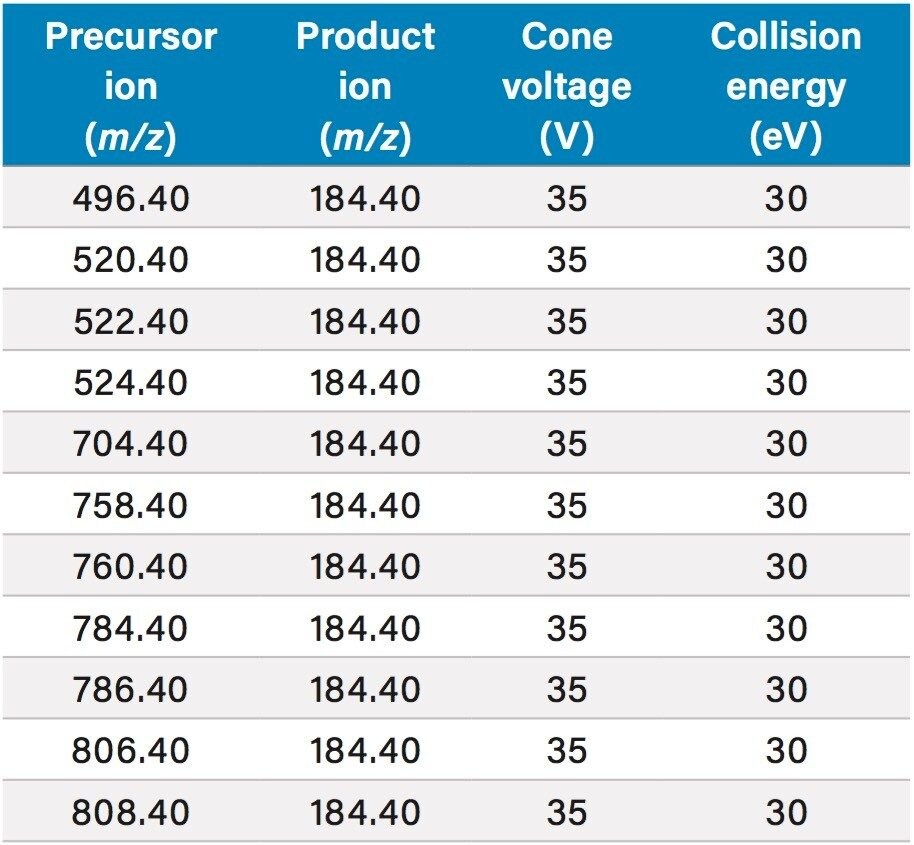
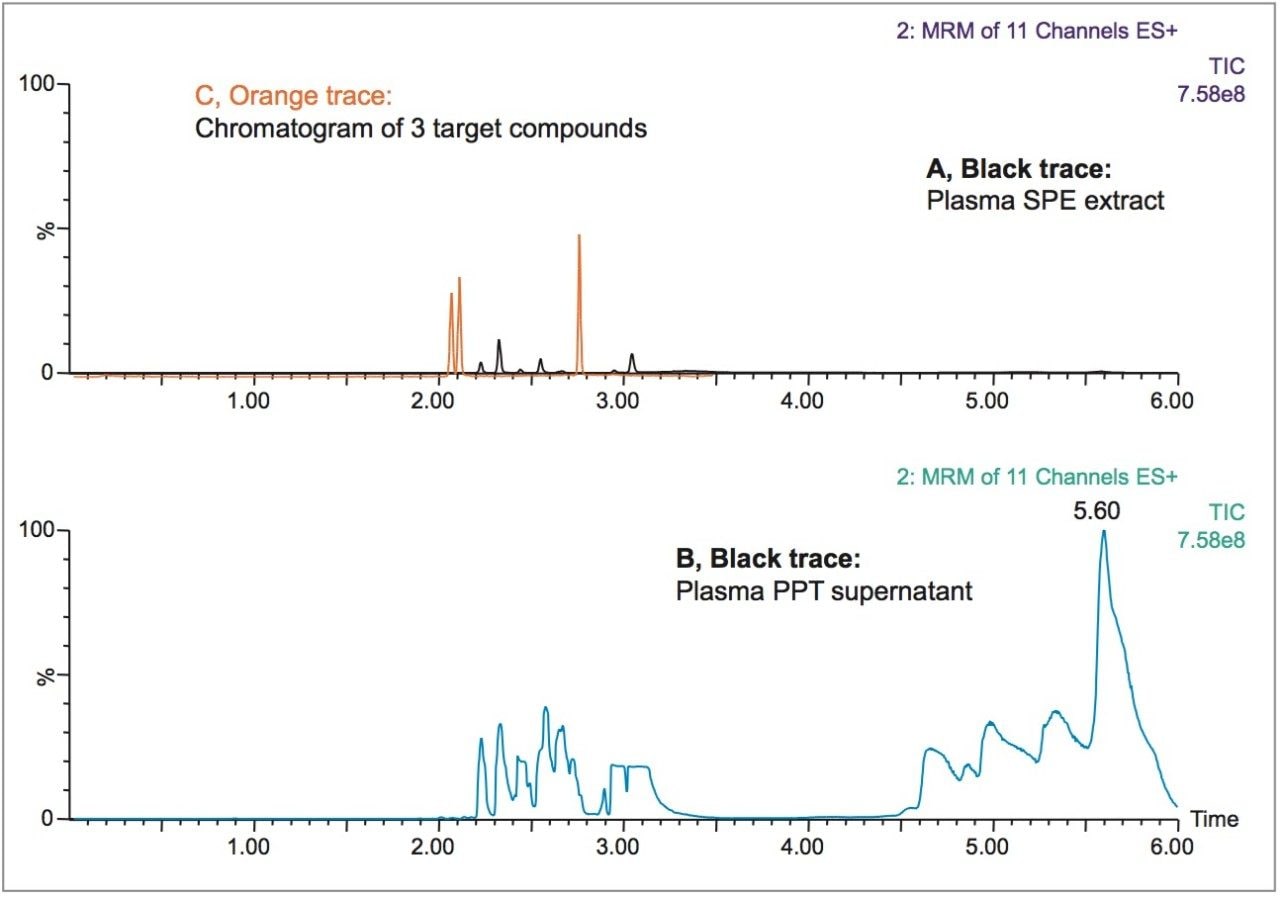
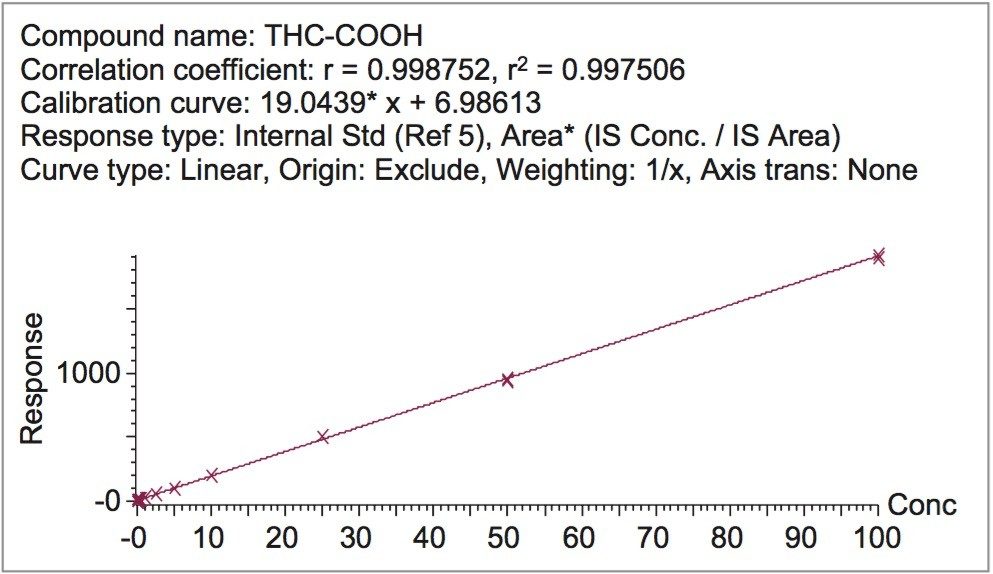
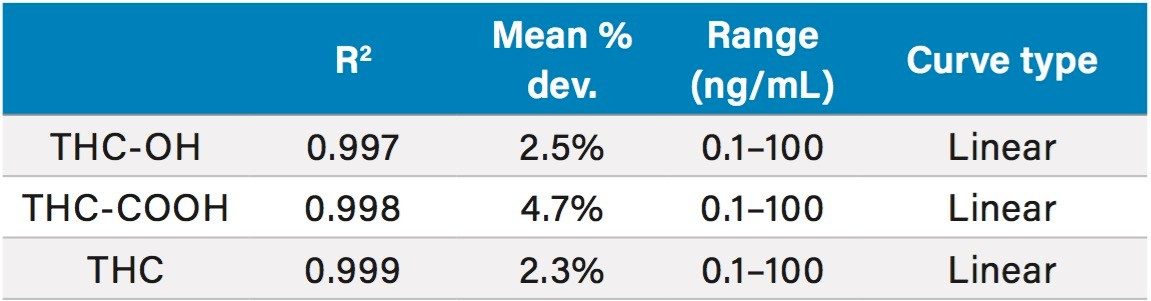
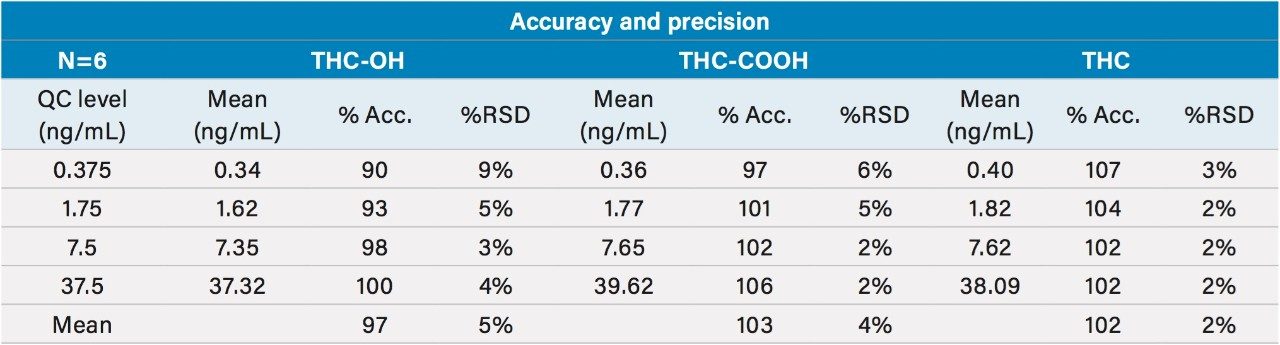
This application details the extraction and analysis of THC and its major metabolites, 11-hydroxy Δ-9-THC (THC-OH) and 11-nor-9-Carboxy-Δ-9-THC (THC-COOH)6 from plasma using an Oasis PRiME HLB µElution Plate, followed by UPLC-MS/MS analysis. The SPE procedure is simple, fast and very efficient, with concentration occurring through the device and elution in LC compatible solvents, allowing for direct injection without subsequent evaporation and reconstitution of samples. Analysis is rapid and highly consistent, with all analytes eluting in less than 3 minutes. Recoveries were excellent (all over 75% with RSDs less than 6%) and matrix effects (ME) were minimal for all compounds (all MEs less than 20%). Quantitative results were highly reproducible. Quality control results were within 10% of expected concentrations and average RSDs within 5%.