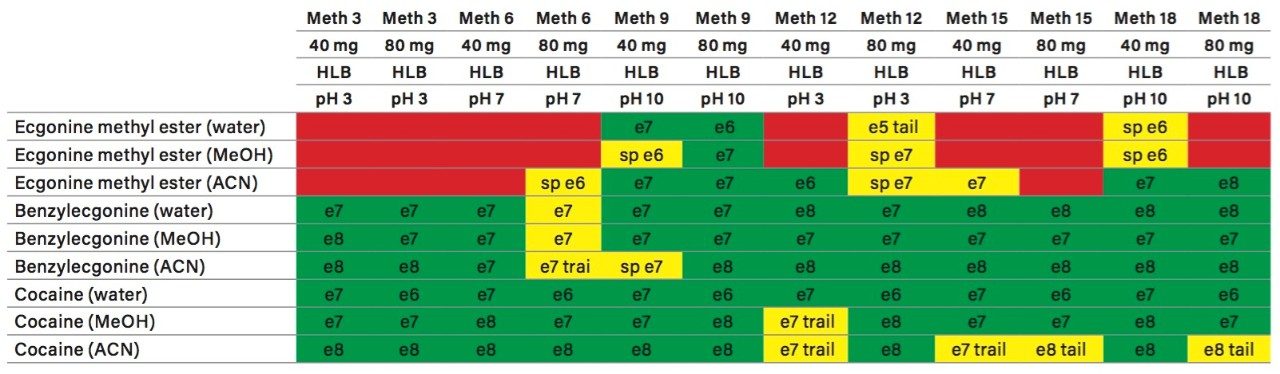
Sample quantification
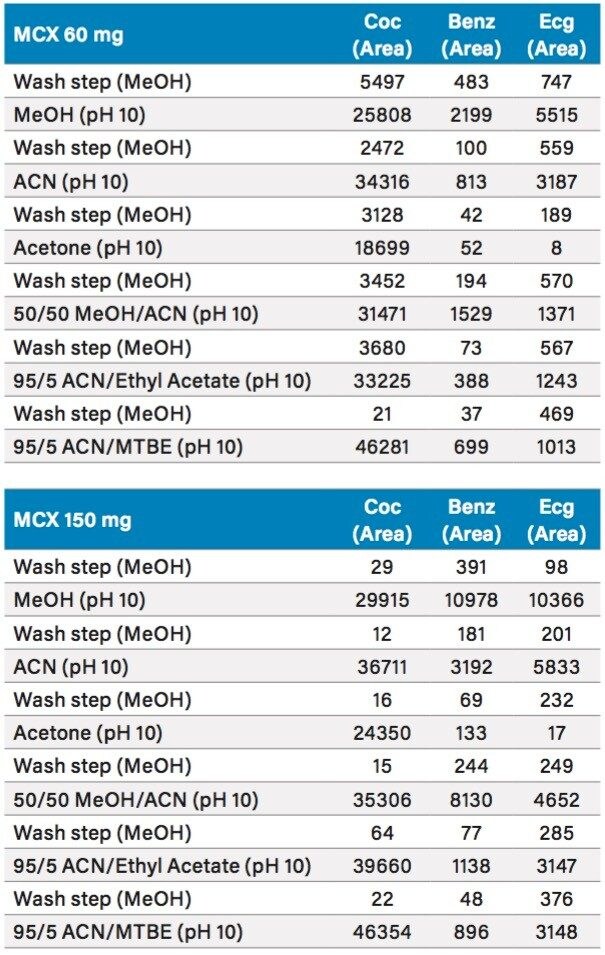
When analyzing highly complex sample types (class C matrix or solids samples), the extraction recoveries are most often overwhelmed by matrix effects, which can lead to either suppression or enhancement in the MS detector. These effects are related to the inability of the extraction protocol to remove interferences from the raw sample. In this work, the extraction protocol relies on a dilution effect (50:1) to avoid the time consuming evaporation to dryness. With the solvent exchange step eliminated, the organic extract from the homogenization process can simply be diluted to reduce the organic content below 5% (optimum value for loading without breakthrough effect during the trapping phase). However, large volume loading will lead to an enrichment effect, which, if poor water quality is utilized, will lead to possible enrichment of additional sources of interferences. For this reason, optima grade water was used for the dilution step. In most cases with complex sample analysis, the use of a deuterated internal standard (post or pre spike) is an effective technique to leverage suppression or enhancement effect from the total recovery. Also, the use of matrix match standards versus neat standards, either extracted or un-extracted, can lead to successful quantification.
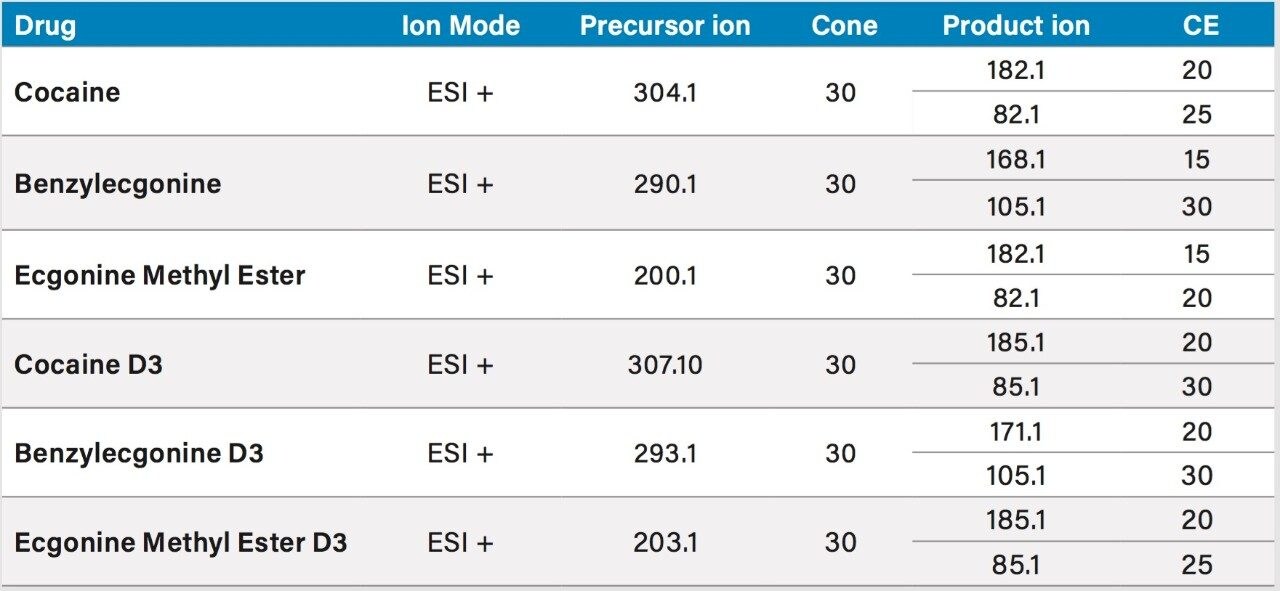
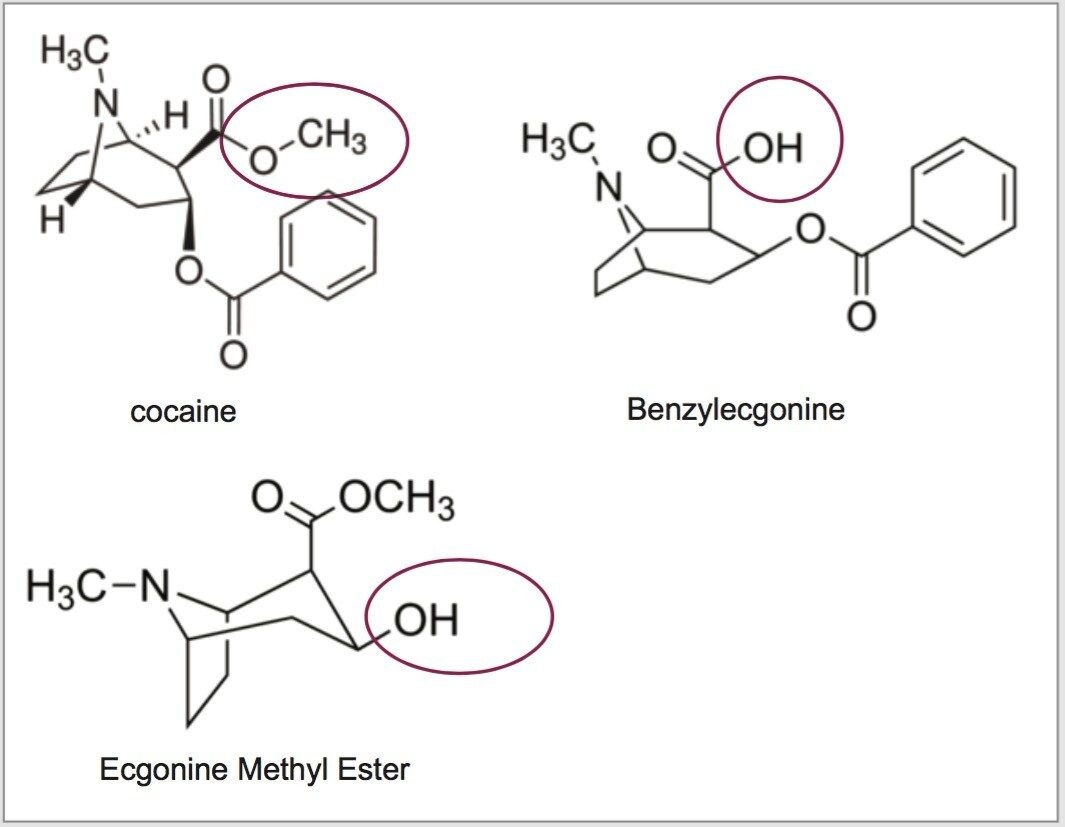
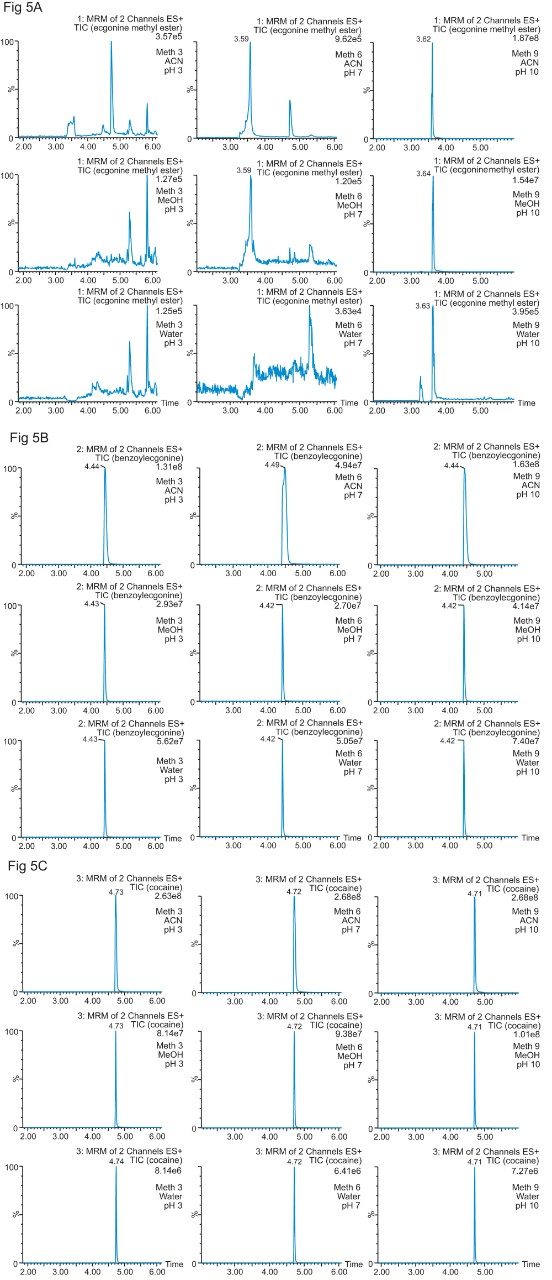
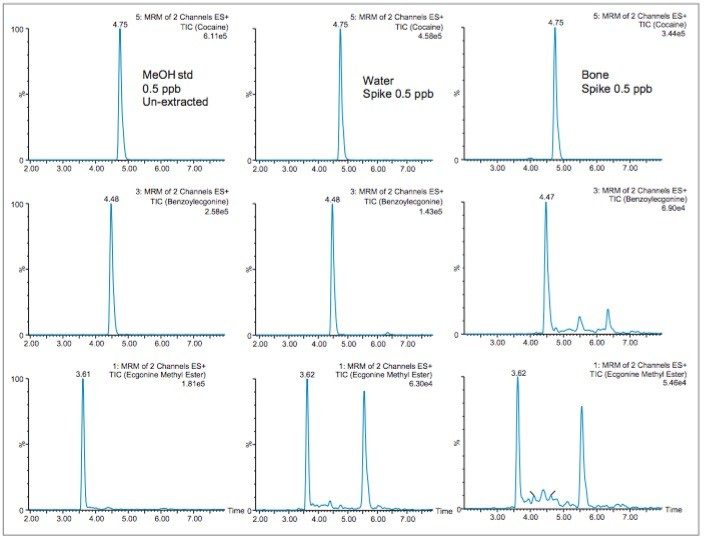
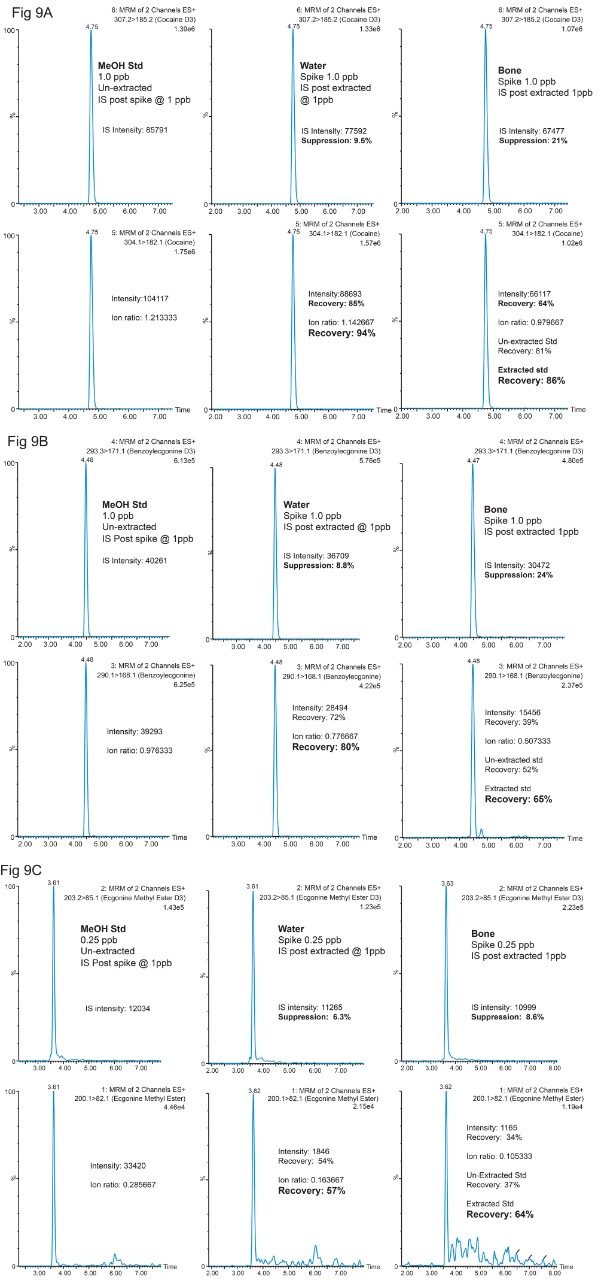
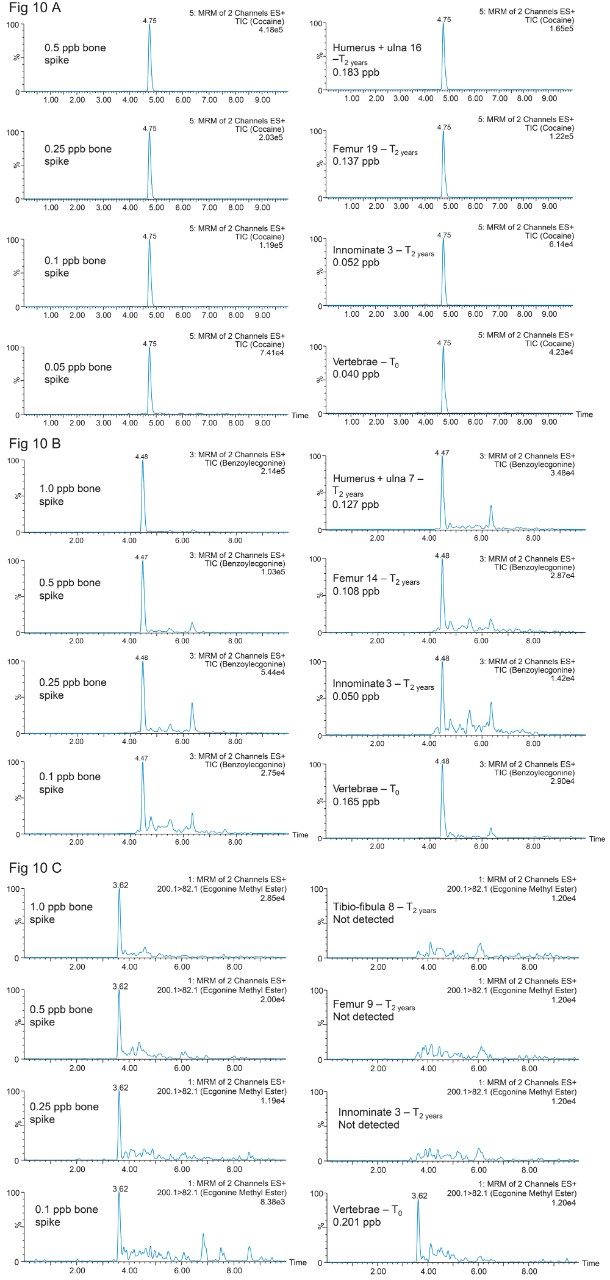
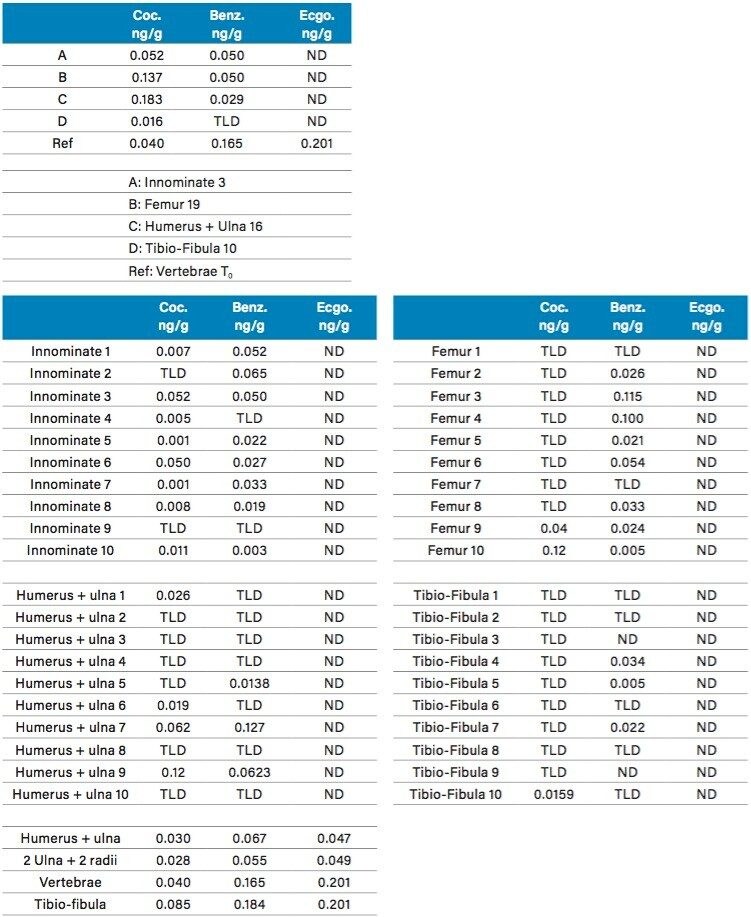
In figure 9A, 9B and 9C, several key chromatograms (neat std, water extract, bone extract) for Cocaine, Benzoylecgonine, Ecgonine methyl ester, showcase the matrix effects and recoveries between neat and extracted standards. For example, in Figure 9A for Cocaine, the bottom left chromatogram shows the response for a cocaine methanol standard at 1 ppb concentration. The deuterated D3 cocaine IS is displayed on the top left chromatogram. From this reference point, a water extracted standard is shown in the bottom middle chromatogram with the corresponding post-spike deuterated IS on top. By comparing the deuterated IS area counts from the neat standard and the post spike extracted standard, the suppression effect for the 100 mL MilliQ enrichment process was calculated at 9.5% for Cocaine, 8.8% for Benzoylecgonine and 6.3% for Ecgonine methyl ester. When using the area counts for recovery calculation for each target analyte, the results show an 85%, 72% and 54%, for Cocaine, Benzoylecgonine, and Ecgonine methyl ester, respectively. These recoveries include matrix effect and potential leachables/extractables from the MilliQ enrichment and the homogenization process. However, by using an ion ratio value (target analyte versus deuterated IS), the recovery value for the extracted water standard shows corrected values for matrix or method effects. Regarding bone analysis, the question concerning quantitation is a challenging one, can a neat extracted standard approach produce acceptable quantifiable results or will the complexity of the sample require a matrix match extracted standards for quantification. In this instance, the latter approach produced satisfactory recovery values in bone matrix of 86% for Cocaine, 65% for Benzoylecgonine and 64% for Ecgonine methyl ester.