In forensic toxicology, drug-screening panels often include commonly used substances such as opiates, benzodiazepines, and stimulants. Often, multiple screening methods are used to obtain a comprehensive view of the multiple drug classes. These methods may include immunoassay, GC-MS, LC-MS/MS, or a combination of methods. Regardless of the methods used, the goal is to achieve sufficient sensitivity, specificity, and accuracy to proceed with the appropriate confirmation, or alternatively, to be confident a sample tests negative.
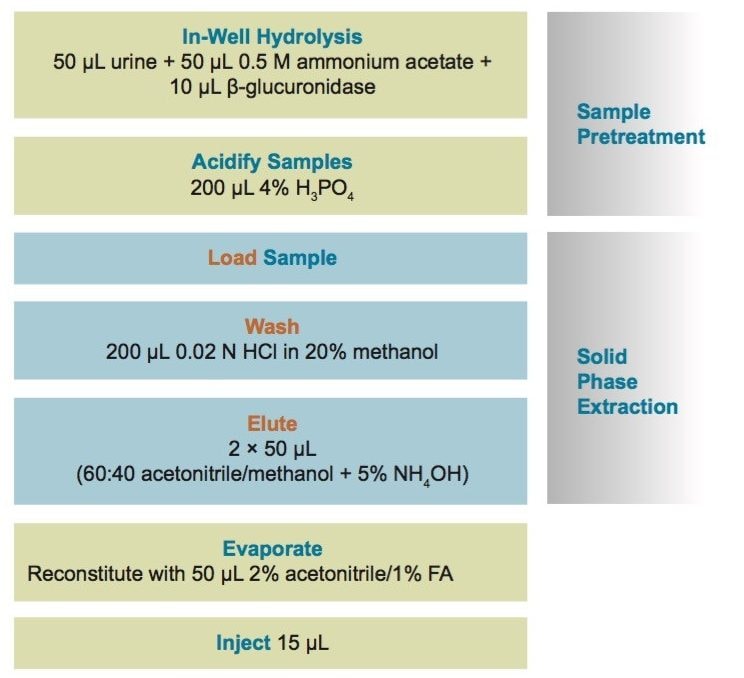
Sample preparation is as important a consideration in forensic toxicology screening as the choice of instrumentation technique. While many laboratories use a “dilute and shoot” approach for urinary toxicology panels, the presence of matrix components, buffers, residual enzymes, and other substances in the sample can result in excessive matrix effects, significantly reduced column lifetimes, and increased instrument downtime resulting from contaminant buildup on electrospray sources in LC-MS.
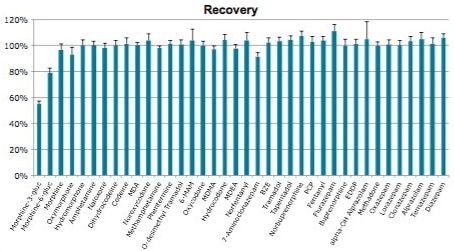
Though solid phase extraction (SPE) is often perceived as difficult or time-consuming, a judicious choice of method can simplify this process significantly. The most selective method of sample preparation, SPE results in cleaner samples than most other techniques, making it ideal for obtaining accurate results.
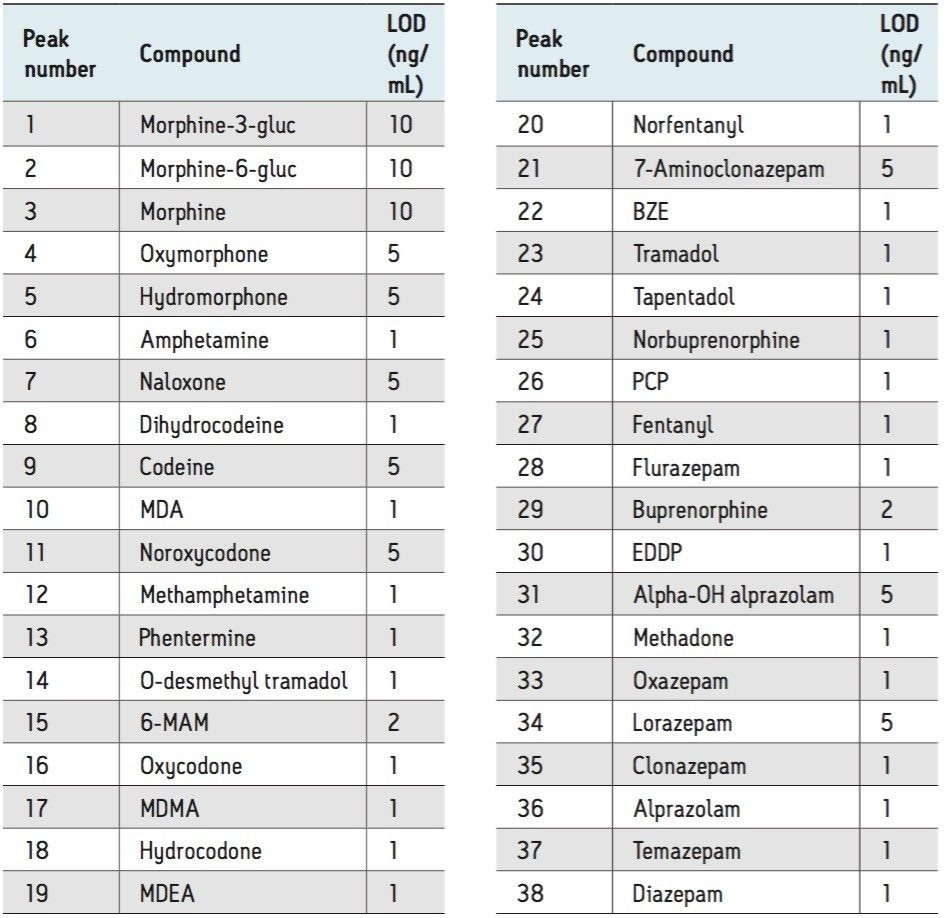
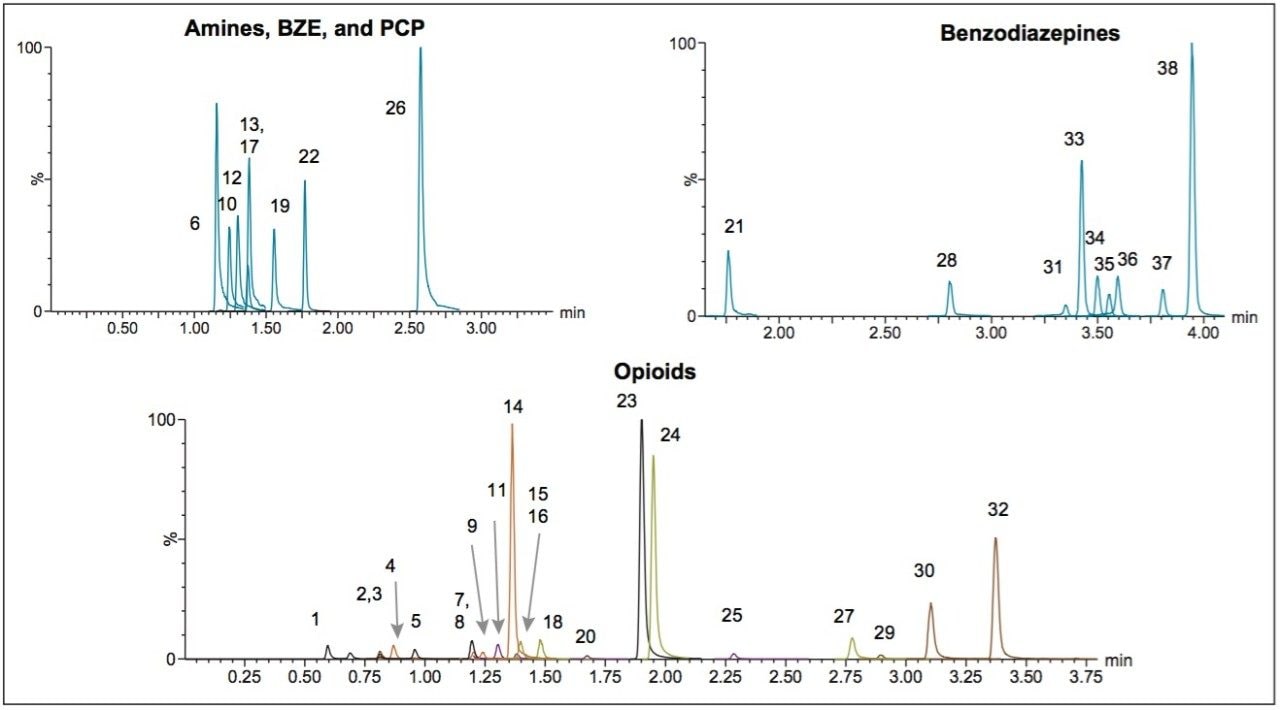
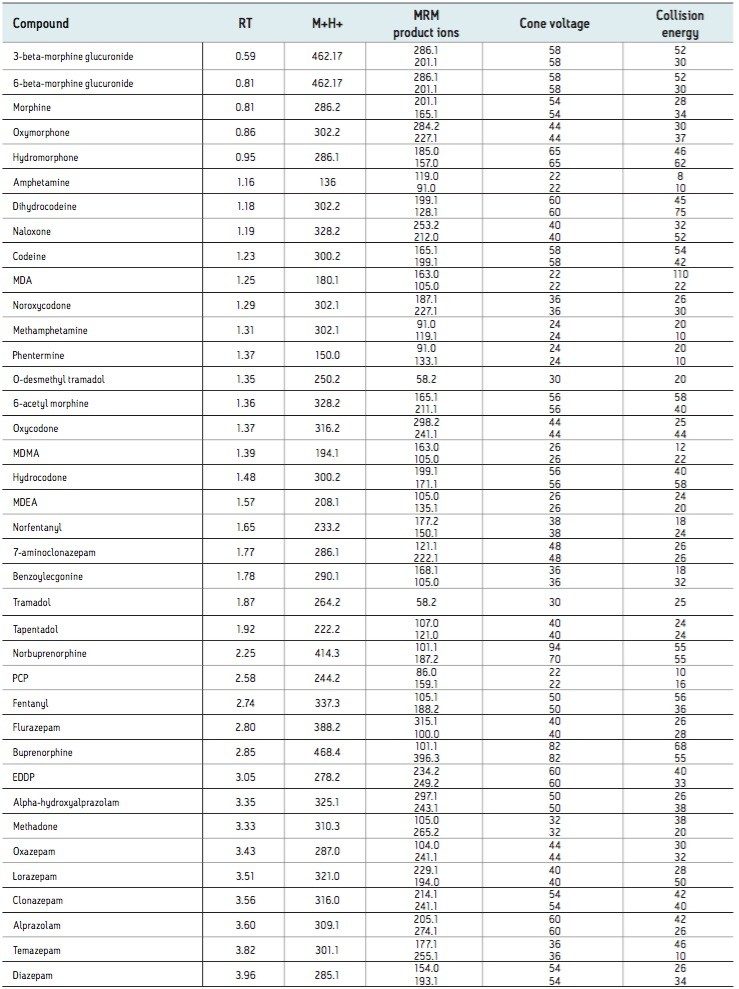
Here we detail a single sample preparation and UPLC-MS/MS analysis strategy for a comprehensive panel of compounds often analyzed in forensic toxicology screens. In an abbreviated, modified extraction method, Waters’ Oasis MCX μElution Plates are used to rapidly extract a panel that includes opioids, amine stimulants, benzodiazepines, benzoylecgonine (BZE), and phencyclidine (PCP). UPLC-MS/MS analysis is achieved using a Waters ACQUITY UPLC BEH Phenyl Column and a Xevo TQD. All sample preparation steps, including enzymatic hydrolysis, are performed within the wells of the micro-elution plates, and the extraction method is simplified, eliminating conditioning and equilibration steps, and consolidating the wash procedures into a single step.